Novel Methods of Necroptosis Inhibition for Spinal Cord Injury Using Translational Research to Limit Secondary Injury and Enhance Endogenous Repair and Regeneration
Article information

Abstract
Spinal cord injuries (SCIs) pose an immense challenge from a clinical perspective as current treatments and interventions have been found to provide marginal improvements in clinical outcome (with varying degrees of success) particularly in areas of motor and autonomic function. In this review, the pathogenesis of SCI will be described, particularly as it relates to the necroptotic pathway which has been implicated in limiting recovery of SCI via its roles in neuronal cell death, glial scarring, inflammation, and axonal demyelination and degeneration. Major mediators of the necroptotic pathway including receptor-interacting protein kinase 1, receptor-interacting protein kinase 3, and mixed-lineage kinase domain-like will be described in detail regarding their role in facilitating necroptosis. Additionally, due to the rapid accumulation of reactive oxygen species and inflammatory markers, the onset of necroptosis can begin within hours following SCI, thus developing therapeutics that readily cross the blood-brain barrier and inhibit necroptosis during these critical periods of inflammation are imperative in preventing irreversible damage. As such, current therapeutic interventions regarding SCI and targeting of the necroptotic pathway will be explored as will discussion of potential future therapeutics that show promise in minimizing long-term or permanent damage to the spinal cord following severe injury.
INTRODUCTION
Spinal cord injuries (SCIs) present with a litany of sequelae and long-term complications including loss of motor function, loss of organ and autonomic function, increased risk of pressure ulcers and pain, and even death [1,2]. Over the last several decades, advancements in approach and treatment of spinal cord injury (SCI) has led to recovery of the aforementioned functions in some cases (though to varying degrees of success) [3-5]. Initially, treatment was designed to attenuate secondary tissue damage as a result of the cascade of pathophysiological processes following the primary damage to neural tissue, which will be described in more detail below [6]. This included pharmacotherapeutic interventions such as corticosteroids which were utilized due to their anti-inflammatory properties and perceived reduction in spinal cord edema, minimizing secondary damage [7,8]. The National Acute Spinal Cord Injury Studies were then conducted to evaluate the efficacy of various doses of methylprednisolone (and later testing the efficacy of the lazaroid tirilazad mesylate as well) in treating SCI [9,10]. Although these studies standardized the use of these pharmacotherapies in the clinical practice of SCI, later criticism of these interpretations included the level of recovery of previously lost motor and sensory functions and potential for further damage as a result of methylprednisolone administration [11-14].
Systemic ganglioside administration of monosialotetrahexosylganglioside (Sygen, Fidia Pharmaceutical Corp., Washington, D.C., USA) has also been proposed as a potential treatment of SCI due to its neuroprotective effects including inhibition of apoptosis and excitotoxicity and increases in neuroplasticity and neurite outgrowth [15-17]. Primary efficacy results could not associate GM1 with marked recovery compared to placebo, however, improvements in motor, sensory, and autonomic function were noted in patients with incomplete paraplegia [18]. Beyond GM1 administration, opioid antagonists have been implicated as a potential therapeutic in SCI to antagonize the rise of endogenous opioids following injury as shown in the literature [19,20]. However, a 5.4-mg/kg intravenous bolus of naloxone followed by a 4-mg/kg 23-hour infusion of naloxone was not found to confer any therapeutic benefit [21]. Additional studies examined the benefit of ion channel antagonists. Calcium channel blockers have been theorized to reduce the pathologic influx of calcium into cells following SCI, while also enhancing blood flow to the spinal cord and reversing hypoperfusion, presenting 3 potential mechanisms of action in the pathophysiology of SCI [22,23]. A therapeutic benefit in a patient population could not be established and risks of systemic hypotension were noted [24]. Finally, cyclooxygenase inhibitors have been investigated as the role of inflammatory prostaglandins mediating secondary injury in SCI can serve as a potential therapeutic target [25]. Studies in animals showed maintenance of blood flow to the spinal following cyclooxygenase-2 inhibition via ibuprofen and meclofenamate, however there is a lack of literature conferring on these findings in humans [26].
Beyond the aforementioned approaches to treating SCI, there are cell therapeutics and nanotechnologies currently being tested with an emphasis on nerve regeneration, including remyelination, axon regeneration, and ultimately the recovery of previously lost nerve function [4,27,28]. One of these approaches involves the use of bone marrow mesenchymal stem cells (BMSCs). Although still in the experimental phase, studies have shown implantation of BMSCs can promote axonal regeneration while limiting immunomodulation, glial scarring, and apoptosis [4,5]. Additional studies are needed regarding long-term complications and efficacy in utilizing these treatments [29-32]. The results of these studies further emphasize the need to investigate alternative molecular pathways and therapeutics to effectively treat, and ultimately cure SCI.
The objective of this study is to analyze novel methods presented in the current literature that is utilized in the inhibition of necroptosis. Further, we wish to investigate the efficacy of these methods in limiting secondary injury.
SPINAL CORD INJURY
1. Mechanism of Action
The most common primary causes of SCI involves the load of a mechanical impact on the spine in which the force of this impact causes disruption and damage to the spinal cord, referred to as impact with either transient or persistent compression (Fig. 1) [6,33-35]. Another primary mechanism of SCI is referred to as distraction which involves stretching and shearing of the spinal column in the axial plane, potentially leading to hemorrhage of the spinal cord vascular supply [35]. Laceration and transection are similar to distraction injuries as they too can lead to hemorrhage of the vascular supply, however in these particular cases, damage typically occurs as a result of sharp fragmentation or severe distraction leading to more significant pathology [35]. This is because laceration and distraction are found to occur with significant trauma and a greater disruption to the vascular supply [35].
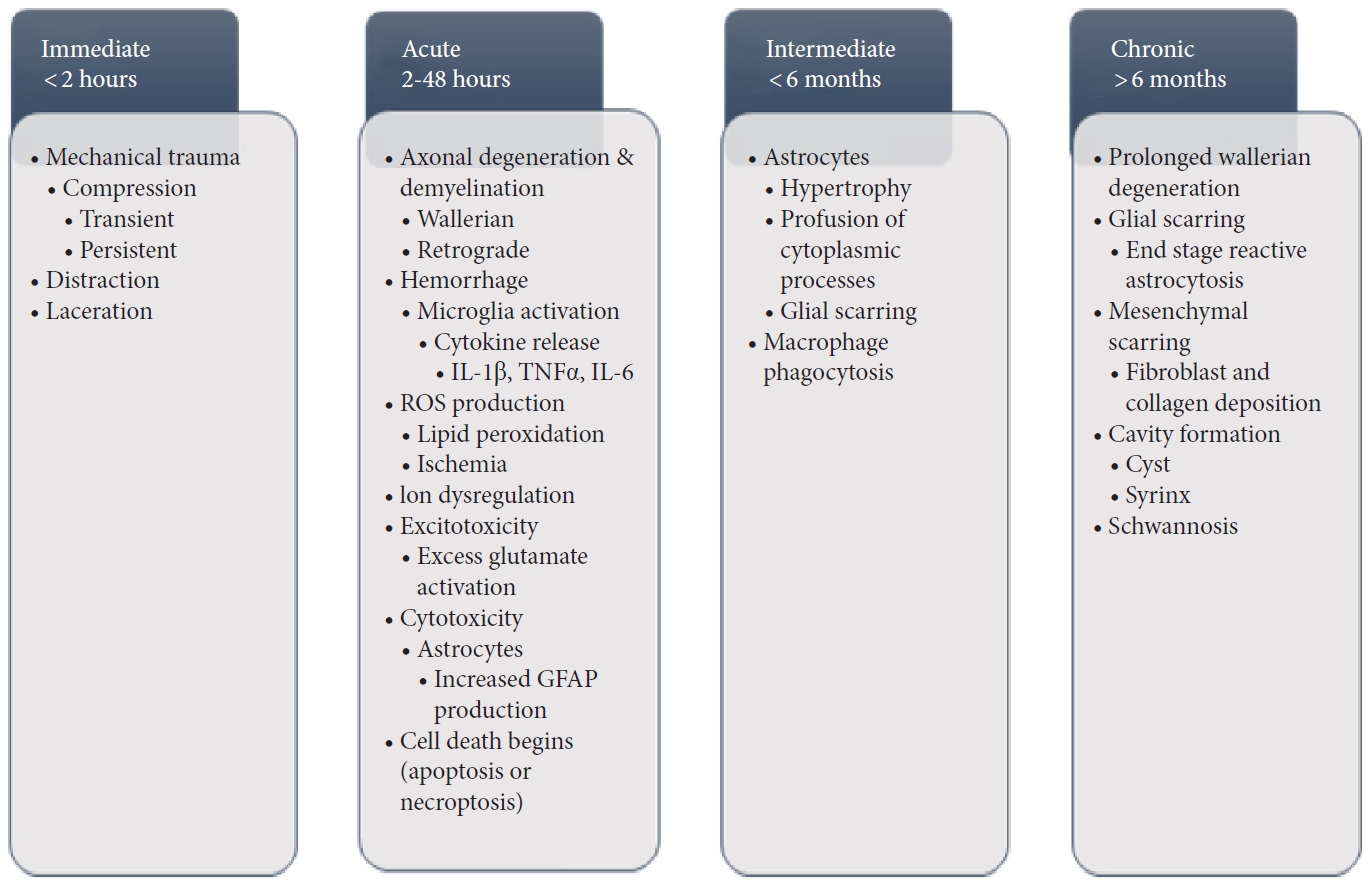
Regardless of the mechanism of primary insult incurred, secondary changes readily follow without immediate intervention which includes axonal degeneration and demyelination that can propagate both in an anterograde (Wallerian) and retrograde manner, affecting both the grey and white matter [33,36,37]. Further, acute hemorrhage can rapidly progress to necrosis of the affected regions, with proinflammatory markers and cytokines including interleukin (IL)-1 beta, tumor necrosis factor-alpha (TNF-α), and IL-6 released at the site of injury via the activation of microglia [34,38]. This is followed by production of reactive oxygen species (ROS), including lipid peroxidation leading to axonal disruption and neuronal/glial death via cell lysis and organelle dysfunction of the aforementioned cells [39,40] and ischemia associated with ROS production [34]. This necrosis and cell death is exacerbated by dysregulation of ionic homeostasis (particularly calcium) and excitotoxicity from excess activation of glutamate receptors at the site of injury [41,42]. Cytotoxicity of astrocytes peripheral to the lesion site confers hypertrophy and proliferation of these astrocytes, leading to an increase in expression of glial fibrillary acidic proteins that coalesce and interweave to form a glial scar which presents as a significant barrier to axonal regeneration and another potential therapeutic target [43-45].
2. Necroptosis
Necroptosis is a caspase-independent process that is instead dependent on receptor-interacting serine/threonine kinase 3 (RIPK3) (described in detail below) in which cellular contents are not neatly packaged into apoptotic bodies [46]. In the case of SCI, studies show that necroptosis presents—along with apoptosis—as the primary forms of programmed cell death in the spinal cord following traumatic injury [47,48]. However, the relationship between these 2 forms of cell death may be more intertwined than initially presumed. Prior studies have described apoptotic death of oligodendrocytes as a result of microglial activation in cases of SCI, as microglial secretion of TNF-α can trigger the apoptotic pathway [49]. However, if upstream signaling is insufficient to trigger apoptosis, TNF-α can instead activate TNF receptor 1 (TNFR1) thus inducing recruitment of receptor-interacting protein kinase 1 (RIPK1) and triggering the necroptotic pathway [48]. Necroptosis can be initiated by a multitude of other signaling pathways as well including death receptors, protein kinase R, DNA-dependent activator of interferon regulatory factors, pattern recognition receptors (PRRs), in addition to TNF signaling which includes RIPK1/3 [50-52]. Cells undergoing necroptosis release their contents into the extracellular space upon death, propagating a proinflammatory state and further cell damage by triggering innate and adaptive immune responses [53,54]. In this proinflammatory state, disruptions in barrier cell integrity allows for microbe invasion leading to recognition of their pathogen-associated molecular patterns (PAMPs) by PRRs that induce the expression of both cytokines and chemokines, including cytokines IL-1α and IL-33, as well as the S100 proteins S100A8, S100A9, and S100A12, which in turn can trigger the death of adjacent cells, propagating a feedback loop that results in non-resolving states of inflammation [54]. In vivo studies show in the necroptotic state there is excessive release of damage-associated molecular patterns (DAMPs) from these dying cells which may consist of cellular organelles and components such as mitochondria or F-actin, HMGB1, as well as nucleic acids, ribonucleoproteins, adenosine triphosphate (ATP), or histone proteins as examples [55]. These DAMPs are recognized by PRRs to induce expression of additional cytokines and chemokines, exacerbating the inflammation initially induced by PAMP recognition [55,56]. Inhibiting this necroptotic inflammatory process may be key to attenuating secondary damage and maximizing therapeutic benefit and recovery by limiting cell death [47,48].
3. Receptor-Interacting Protein Kinase 1
RIPK1 is an important effector downstream of death receptors and PRRs that govern prosurvival, apoptotic, and especially, inflammatory necroptotic pathways. Thus, much interest has focused on therapeutically targeting RIPK1 in the context of numerous inflammatory diseases. Multiple small molecule inhibitors of RIPK1 have demonstrated protective effects in mouse models of autoimmune or inflammatory disease. These studies were primarily conducted using molecular analogues of necrostatin-1 (Nec-1), which was originally identified in a chemical compound screen. Since then, other chemical families have been identified to inhibit RIPK1, several of which have excellent blood-brain barrier permeability, offering opportunities to address neuroinflammatory central nervous system (CNS) conditions [47]. Several of these inhibitors are currently in phase I and/or II clinical trials by Denali Therapeutics and GlaxoSmithKline, primarily for the treatment of Alzheimer disease (AD) and amyotrophic lateral sclerosis (ALS) [48]. Notably, the compound DNL788, a novel brain-penetrant RIPK1 inhibitor, was recently announced by Denali in partnership with the Sanofi biopharmaceutical company to initiate clinical testing in 20201 for neurodegenerative indications [49]. Combination therapy with RIPK1 inhibitors is also feasible and has, in several instances, demonstrated benefit [50,51]. For example, Cougnoux et al. [51] showed that treating mouse models of Niemann-Pick disease type C1 with a combination of the RIPK1 inhibitor GSK547 and the compound HPβCD, which slows neurological decay, resulted in delayed loss of Purkinje neuron density. The neuroprotective value of combination therapies involving RIPK1 inhibitors has not been extensively evaluated in a clinical setting yet represent a promising practical approach toward limiting neurological damage, such as in SCI.
On a molecular level, the activation of RIPK1 has been most studied in the context of TNF signaling via its activation of TNFR1. Binding of TNFR1 to TNF leads to recruitment of RIPK1 via its death domain, which results in the activation of the proapoptotic caspase-8. Typically, caspase-8 inhibits RIPK1-mediated necroptosis, resulting in apoptotic progression; however, when caspase-8 is inhibited, RIPK1 recruits RIPK3 and mixed-lineage kinase domain-like (MLKL), leading to necroptosis [52]. Under in vitro conditions, necroptosis is typically elicited artificially by treating cells with a combination of TNF and a pan-caspase inhibitor, which raises the question as to in what in vivo contexts necroptosis plays a role. Despite this, studies have demonstrated that abrogation of RIPK1 kinase activity, either pharmacologically or genetically, in mouse models of AD, ALS, and multiple sclerosis (MS) were neuroprotective [53]. In the context of SCI, the role of RIPK1 has not been as comprehensively elucidated. However, given the shared pathophysiological mechanisms between SCI and other neurodegenerative diseases, targeting RIPK1 may prove effective.
Several lines of evidence strongly implicate necroptosis and RIPK1 in the pathogenesis of SCI. First, in the context of MS and ALS, RIPK1 has been shown to contribute to oligodendrocyte dysfunction, causing axonal demyelination [53]. Interestingly, oligodendrocytes are one of the few cell-types that engage necroptosis downstream of TNFR1 signaling without the necessity of caspase inhibition, suggesting these cells may be inherently primed to engage the inflammatory necroptotic pathway [54]. In the context of SCI, oligodendrocyte necroptosis and death impairs axonal function and exacerbates pathology [55]. Therefore, beyond MS and ALS, RIPK1 inhibition may reduce oligodendrocyte dysfunction and improve axonal survival following SCI. Second, apart from inducing cell death, RIPK1 activation also promotes the production of proinflammatory cytokines, notably by myeloid cells such as CNS-resident microglia. In SCI, microglia have been posited to contribute to various aspects of pathogenesis, however, these findings are complicated by (1) the involvement of both microglia and its related myeloid cell-type, monocyte-derived macrophages (MDMs), and (2) the dual capacity for microglia to be neurotoxic and neuroprotective [56]. Despite this, since RIPK1 is critically involved in the inflammatory, neurotoxic activities of both microglia and MDMs in CNS disease, it remains a target with therapeutic promise. Accordingly, Fan et al. [57] in 2015 showed that Nec-1 treatment of mice with SCI reduced the SCI-induced increase in microglia/macrophage cell death. Lastly, RIPK1-mediated necroptosis in astrocytes has also been shown to contribute to SCI. Generally, astrocytes have protective, neurotrophic roles in SCI [58,59]. In a separate publication, Fan et al. [60] also showed that microglia/macrophages in SCI can induce astrocyte necroptosis, diminishing their neuroprotective effects. In SCI mice, depletion of microglia resulted in higher numbers of live astrocytes and treatment with Nec-1 decreases astrocyte necroptosis as well as increases neuronal cell number. Generally, 3 studies have most directly evaluated the potential of RIPK1 inhibition, specifically with Nec-1, as a treatment for SCI [61-63]. The treated SCI mice with Nec-1 and demonstrated a reduction in neuronal death and grey matter lesion area [61-63]. More detailed inspections of post-SCI Nec1-treated neurons showed reduced apoptosis, necroptosis, and oxidative stress as well as improved mitochondrial function [61-63]. From a behavioral standpoint, Nec-1-treated mice also displayed quicker motor recovery and better open-field mobility following recovery [61-63]. Overall, RIPK1 exhibits pleiotropic effects contributing to the exacerbation of SCI and sufficient evidence supports the therapeutic utility of RIPK1 inhibitors.
4. Receptor-Interacting Protein Kinase 3
Receptor-interacting protein kinase 3 (RIPK3) is a member of the RIP family. Similar to RIPK1, RIPK3 can trigger necrosis independently. However, most of its known functions have been studied when its works in conjunction with RIPK1 [64]. RIPK1 and RIPK3 interact with each other via the RIP homotypic interaction motif which leads to formation of the necrosome that activates downstream effector proteins to elicit the above-mentioned necroptosis pathway and inflammatory response [65]. In mice, RIPK3 expression is elevated just 24 hours after spinal cord hemisection [66]. An increase in RIPK3 has been shown to contribute to cell loss via its necroptotic pathway. This is the fundamental component leading to neurodegenerative diseases in SCI patients [67]. In addition to its necroptotic properties, RIPK3 can activate caspase-independent cell death through TNF-induced mitochondrial generation of ROS [68]. This increase in ROS is not only correlated with cell death but works in a positive feedback loop to enhance necrosome formation and necroptosis.
The protease caspase-8 and IAP ubiquitin ligases inhibit RIPK1/RIPK3 oligomerization, signaling and thus prevents necroptosis (Fig. 2) [69]. Inhibitors of these 2 have been used to study necroptosis for years since they inhibit apoptosis and trigger necroptosis. However, clinically neither of these proteins have been successfully targeted for treatment. Nevertheless, the search for RIPK3 specific inhibitors has been an area of ongoing research. Recent studies suggest that RIPK3 inhibitor, GSK872, improves motor function and spinal cord edema in a SCI mouse model [70]. GSK872 is part of a group of kinase inhibitors. These inhibitors have a type I, II, or III kinase binding mode, with type I binding the ATP-binding site, type II interacting with the hinge region of ATP-binding site, and type III binding the inactive hydrophobic back pocket of the kinase domain [71]. Very few of these inhibitors have been successfully selected for the treatment of disease; having most of their use in cancers. A better understanding of the kinome selectivity and specificity along with an increase in in vivo testing of these drugs can help us move towards faster clinical implementation of RIPK3 inhibitors.
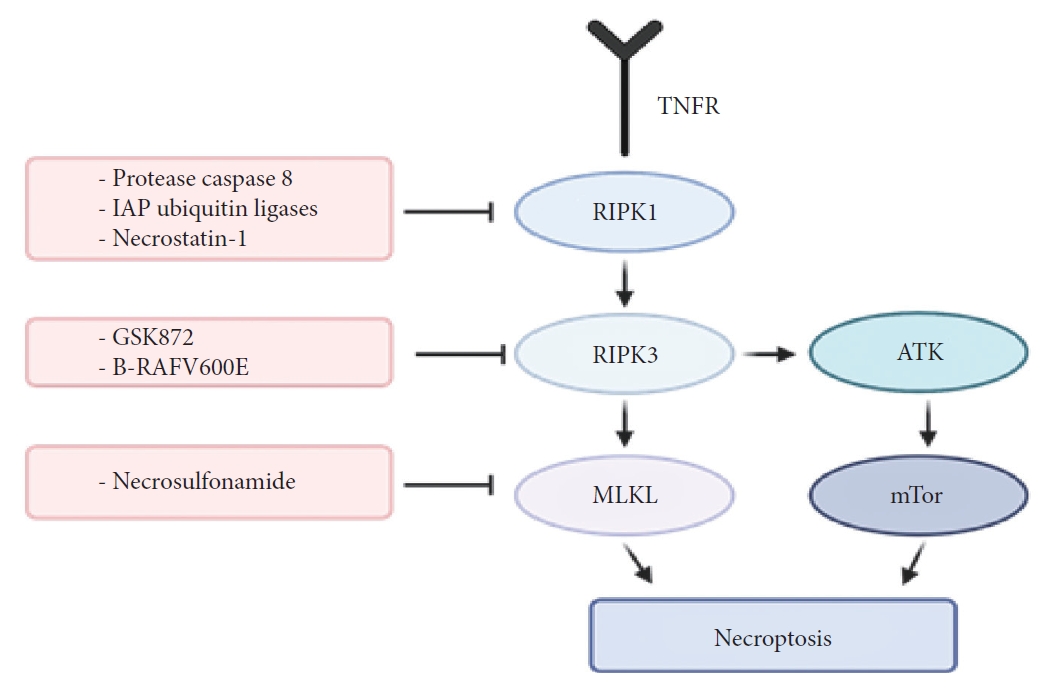
The action mechanism of RIPK1, RIPK3, MLKL, and combination of necroptosis inhibition with PI3K/AKT/mTOR pathway inhibition. RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; MLKL, mixed-lineage kinase domain-like; PI3K, phosphatidylinositol 3 kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; IAP, inhibitor of apoptosis protein; TNFR, tumor necrosis factor receptor.
Although TNF death receptor, caspase-8, RIP1, and RIP3 are the most studied and important molecules that regulate cell apoptosis and necroptosis, the innate immune system has a set of pathogen-associated receptors that can also lead to cell death. Necroptosis can be triggered by PRRs. These are proteins capable of detecting conserved microbial products and endogenous damaged molecules. There are 4 major subfamilies of PRRs—the Toll-like receptors (TLRs), the nucleotide-binding oligomerization domain–leucin rich repeats-containing receptors, the retinoic acid-inducible gene 1 (RIG-1)-like receptors, and the C-type lectin receptors. There is evidence that 2 of the 13 TLRs and intracellular sensing proteins, such as RIG can lead to necroptosis. Most endosomal and plasma-membrane associated TLR respond to pathogens and induce necrosis partially through TNF and RIP activation [72].
MIXED-LINEAGE KINASE DOMAIN-LIKE PROTEIN
As previously alluded to, necroptosis is initiated by TNF and the activity of RIP1 and RIP3. However, another important factor that mediates the activation of necroptosis is MLKL [73,74]. MLKL is another mitochondrial protein that serves as a substrate for RIP3 [75]. The RIP1/RIP3 complex initiates and activates programmed necrosis after injury, secondary to phosphorylation of MLKL, thereby causing mitochondrial dysfunction. Given the important role of MLKL, several studies have investigated the effect of manipulation of this factor and the associated pathway. Jiao et al. [76] in a recent study used necrosulfonamide (NSA) to block MLKL, as means to prevent mitochondrial dysfunction. Their results showed that blocking MLKL using NSA prevented a decrease in mitochondrial membrane potential, ATP, glutathione, and superoxide dismutase levels and also prevented an increase in ROS and malondialdehyde levels. In terms of functional effects, the authors showed that among mice treated with NSA to block MLKL, there was a significant improvement in locomotor function [76]. The authors also demonstrated an optimal therapeutic window for treatment with NSA to block MLKL, which was within the first 12 hours of injury. These results show that blocking MLKL may provide an effective way of preventing secondary injury after SCI.
COMBINATION OF NECROPTOSIS INHIBITION WITH PI3K/AKT/mTOR PATHWAY INHIBITION
It has previously been demonstrated that following SCI, RIPK1 and RIPK3 mediate necroptosis, which in addition to several pathways, also involves inhibition of autophagy [77]. Autophagy is a catabolic pathway which has been shown to facilitate degradation of cytoplasmic content in a lysosome-dependent manner [78]. The autophagic flux, consisting of autophagosome formation, maturation, fusion with lysosomes, subsequent breakdown, and the release of macromolecules back into the cytosol, is mediated by several other molecules called the autophagy-related (ATG) protein family. Several studies have suggested a neuroprotective effect of autophagy after traumatic brain injury, including preservation of neurobehavioral function, increased neuronal survival, reduced inflammation and gliosis in the injured brain, and preventing further cell death and apoptosis [79-81]. One of the most significant mediators of autophagy is the phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway [82]. Among these, mTORC1, a component of mTOR has been shown to be a negative mediator of autophagy [83-85]. while PI3K/AKT, in turn, modulates mTORC1 [86]. Therefore, combining RIPK1/RIPK3 inhibition with inhibition of mTOR (for e.g., with rapamycin) may help to simultaneously activate autophagy while also inhibit activation of necroptosis pathway, thereby preventing further cell death.
FUTURE PERSPECTIVES
Current therapeutics directed towards specifically inhibiting MLKL are limited. One of the most promising candidates includes the chemical NSA which has shown the ability to attenuate necroptosis in SCI, however additional studies are needed to validate these findings which have only been described in one study thus far [87].
CONCLUSION
Spinal cord injuries present with a complex series of molecular cascades that ultimately induce cell death of neurons and glia and excitotoxicity of astrocytes, limiting the effect of therapeutic intervention in these damaged regions. In this review, the mechanisms of action underlying SCI were discussed, particularly the roles of RIPK1/RIPK3 signaling pathways and the induction of necroptosis via the activation of death receptor ligands and caspase inhibition. Further emphasis was placed on potential therapeutics to limit the degree of necroptosis, in particular inhibition of RIPK1/3 and mTOR to increase rates of autophagy while inhibiting the necroptotic pathway in order to preserve cell survival and promote recovery via reduced inflammation, gliosis, and cell death.
Additional studies are necessary to investigate and develop therapeutics that successfully inhibit the necroptotic pathway and facilitate recovery following SCI. Studies on the RIPK1 inhibitor Nec-1 implicate this drug as a potential therapeutic as it is highly permeable across the blood-brain barrier with minimal neurotoxicity while ultimately limiting neuronal cell death. Currently, there are 6 human clinical trials looking at RIPK1 inhibitors in ALS, AD, psoriasis, ulcerative colitis, rheumatoid arthritis, and pancreatic ductal adenocarcinoma. Further, selective inhibition of RIPK3 via administration of B-RAFV600E inhibitor dabrafenib may be a potential focus of investigation. With the potential of concomitant RIPK1 and RIPK3 inhibition via coadministration of these therapeutics, there holds great promise in sufficient inhibition of the necroptotic pathway following SCI. However, the combined pharmacological effects of these therapeutics have yet to be explored following coadministration. Additionally, these interventions have not been sufficiently studied in human models of SCI either, warranting further investigation in that regard.
Notes
The authors have nothing to disclose.