Gene Therapy Approach for Intervertebral Disc Degeneration: An Update
Article information

Abstract
Intervertebral disc degeneration is the primary cause of back pain and associated with neurological disorders including radiculopathy, myelopathy, and paralysis. The currently available surgical treatments predominantly include the excision of pathological discs, resulting in the function loss, immobilization, and potential additional complications due to the altered biomechanics. Gene therapy approach involves gene transfer into cells, affects RNA and protein synthesis of the encoded genes in the recipient cells, and facilitates biological treatment. Relatively long-exerting therapeutic effects by gene therapy are potentially advantageous to treat slow progressive degenerative disc disease. In gene therapy, the delivery method and selection of target gene(s) are essential. Although gene therapy was first mediated by viral vectors, technological progress has enabled to apply nonviral vectors and polyplex micelles for the disc. While RNA interference successfully provides specific downregulation of multiple genes in the disc, clustered regularly interspaced short palindromic repeats (CRISPR) system has increased attention to alter the process of intervertebral disc degeneration. Then, more recent findings of our studies have suggested autophagy, the intracellular self-digestion, and recycling system under the negative regulation by the mammalian target of rapamycin (mTOR), as a gene therapy target in the disc. Here we briefly review backgrounds and applications of gene therapy for the disc, introducing strategies of autophagy and mTOR signaling modulation through selective RNA interference.
INTRODUCTION
1. Socioeconomic Backgrounds and Clinical Symptoms of Intervertebral Disc Disease
Back pain increases with age, affects 70–85% of people during their lives [1], and causes disability with estimated healthcare costs up to $100 billion/year in the US [2]. The cause of back pain is multifactorial; however, intervertebral disc degeneration has been revealed to be one of the independent risk factors [3]. Furthermore, disc degeneration can present not only back pain but also radicular pain, numbness, muscle weakness, and then paralysis in the worst scenario [4]. Intervertebral disc herniation, an injury with degenerative wear and tear, occurs even in the youth, limits daily and sports activities, and requires surgery in 10% [5]. Lumbar spinal stenosis, a clinical condition narrowing the spinal canal and/or intervertebral foramina, arises in conjunction with age-associated degeneration of the lumbar discs and facet joints, resulting in the most frequent indication of spinal surgery in elderly patients [6].
2. Pathophysiology of the Intervertebral Disc
The intervertebral disc has a complex structure with the nucleus pulposus (NP) encapsulated by the annulus fibrosus (AF) and endplates [4]. The collagenous, laminar AF surrounds the central, gelatinous NP, maintaining pressurization of the NP for support of compressive loading and facilitation of multiaxial spinal movement [4]. While the AF comes from the mesenchyme [7,8], the origin of the NP is the notochord [9]. Notochordal cells only exist during the first decade of human life and are subsequently replaced by non-notochordal, chondrocyte-like cells of unknown provenance [7,8]. Disc cells have a chondrocytic phenotype to produce extracellular matrix components comprising proteoglycans (principally aggrecan) and collagens (primarily types I in the AF and II in the NP) [10]. Matrix metabolism is maintained by catabolic enzymes, matrix metalloproteinases (MMPs) and disintegrins and metalloproteinases with thrombospondin motifs (ADAMTSs), and anticatabolic inhibitors, such as tissue inhibitors of metalloproteinases (TIMPs) [11]. Increased expression of MMPs and ADADAMTSs relative to TIMP has been observed during clinical human [12-14] and experimental rodent disc degeneration [15-17]. The disc is an immune-privileged, the largest avascular organ in the body [18,19]. Disc cells thus live under an extremely harsh environment—low glucose, oxygen, and pH and high osmolality and load fluctuation [20]. The nutrient supply of central disc NP cells depends on the diffusion from blood vessels at the disc margins compared to peripheral disc AF cells [19]. Therefore, decreased blood supplies, subchondral bone sclerosis, and endplate calcification, caused by mechanical stress, injury, smoking, and aging, can be harmful for the transport of nutrients to discs and are suspected to contribute to the development of disc degeneration [19].
In humans, age-related disc changes characterized by the loss of notochordal cells start from early childhood as observed in people aged 11–16 years [21,22], ~40% of people aged under 30 years and 90% of those aged over 55 years present structural lumbar disc degeneration [23]. Cell decrease is another major characteristic of disc degeneration and primarily results from programmed cell death, apoptosis [24]. Apoptotic cells increase substantially between the ages of 11–16 years, associated with the loss of notochordal cells and chondrocyte proliferation [22], suggesting a relationship between apoptosis and notochordal cell disappearance during the pathogenesis of disc degeneration [7]. Furthermore, a notably high incidence of apoptosis has been observed during human [25] and rodent disc aging and degeneration [17,26]. The incidence of irreversible cell growth arrest due to aging, senescence [27], also increases during human disc degeneration [28,29]. In addition, we have more recently focused on autophagy as a potent treatment target for human degenerative disc disease [30,31]. Autophagy, the intracellular self-digesting and recycling process, is an important cell survival mechanism that sustains metabolism and prevents the accumulation of toxic proteins and organelles under stress conditions, especially nutrient deprivation [29,32-34]. Age-dependent and degeneration severity-dependent changes in the involvement of autophagy-related genes and proteins have been found in human disc specimens surgically collected [30,31]. Understanding of the unique environment, cell fate, and matrix metabolism in the disc is required to design new biological therapies for degenerative disease.
3. Current and Future Treatment of Intervertebral Disc Degeneration
It is difficult to reverse, stop, or even delay the degenerative process by using the currently available treatment options, because of the limited regenerative potential of intervertebral disc tissues [35,36]. Despite successful conservative treatment outcomes with medication and physiotherapy in disc disease [37], nonresponders reluctantly require surgery [5]. Surgical interventions predominantly include the excision of pathological discs, resulting in the function loss, immobilization, and potential additional complications due to the altered biomechanics [38]. Therefore, the development of biological approaches for disc regeneration is an urgent issue.
Biological therapies for intervertebral disc degeneration can be divided into 3 major groups: growth factor injection with or without a carrier [39], cell-based therapy with or without a scaffold [40], and gene therapy modifying endogenous gene expression and function [41]. This review article focuses on backgrounds and applications of gene therapy in the treatment of spinal disorders, especially associated with degenerative disc disease (Table 1).
Types of gene therapy for degenerative intervertebral disc disease
DATA SOURSES AND SEARCH
We searched the keywords of “gene therapy” and “intervertebral disc” in PubMed (https://pubmed.ncbi.nlm.nih.gov/) from January 1997 to December 2019 and reviewed the abstracts and full-text articles. We also searched the keywords of “RNA interference,” “autophagy,” and “CRISPR.”
GENE THERAPY
1. Mechanism and History of Gene Therapy
Gene therapy is defined as the transfer of either RNA or DNA to treat or prevent a disease. Targeted diseases have been mainly classic and fatal genetic disorders, however, the application of gene therapy for such diseases as acquired chronic disorders has been allowed owing to technical advances. A major advantage of gene therapy is its relatively long-term efficacy. Once a therapeutic gene is successfully transferred into the target cells, these genetically modified cells continue to produce the desired gene products (RNAs or proteins). Disc degeneration and related disorders are chronic conditions and therefore good candidates for gene therapy.
In vitro gene therapy for disc degeneration was first reported in 1997, which transferred genes to chondrocytic cells of bovine endplates using retroviral vectors [42]. In vivo gene therapy for disc degeneration was first reported in 1998, which achieved sustained expression of β-galactosidase (lacZ) with adenoviral vectors in a rabbit model at 1 year [43]. Thereafter, a variety of therapeutic genes have been studied and delivered to disc cells via viral and nonviral vectors [41]. These vectors are required to transfer the gene of interest into the target cells, because genetic materials are not well transmitted into cells. Viral vectors have been preferred since they can enter into cells and subsequently express their genes as a part of the life cycle, resulting in greater long-term transgene expression.
Another essential factor in gene therapy is how to deliver the genetic materials to the target organ. Because the intervertebral disc is avascular and encapsulated, and the intradiscal environment is harsh for cells to survive, local and direct gene therapy is the best way for the disc, which gene of interest is administered locally and delivered directly to the cells of the target organ in vivo [41].
2. Virus-Mediated Gene Therapy
1) Retrovirus
Wehling et al. [42] reported the in vitro transfer of 2 different exogenous genes via retroviral vectors to chondrocytic cells isolated from bovine coccygeal vertebral endplate. The bacterial LacZ gene and, alternatively, the complementary DNA (cDNA) of the human interleukin-1 (IL-1) receptor antagonist were introduced into the chondrocytic cells. Transfer of the LacZ gene to cultured cells resulted in ~1% LacZ positive cells by 5-bromo4-chloro-3-indolyl-β-galactosidase (X-Gal) staining, and transfer of IL-1 receptor antagonist cDNA resulted in the production of 24 ng/mL/106 cells IL-1 receptor antagonist protein in 48 hours by enzyme-linked immunosorbent assay. This study indicated the potential use of local gene therapy for disc degeneration.
2) Adenovirus
Nishida et al. [43] reported the in vitro and in vivo transfer of the lacZ receptor gene to rabbit disc cells. In vitro, disc NP cells isolated from skeletally mature female New Zealand white rabbits were cultured and transfected with adenovirus constructs encoding the lacZ gene (Ad-lacZ). In vivo, Ad-lacZ in saline solution was injected directly into the NP of lumbar intervertebral discs of the rabbits by anterior approach. In vitro and in vivo X-Gal staining showed efficient transduction of disc NP cells, and reporter gene expression was maintained in vivo for at least 12 weeks. This study demonstrated that exogenous genes were successfully transferred to the disc in vitro and in vivo via adenoviral vectors, which indicated the feasibility of direct gene therapy for spinal disorders.
In the following study, adenovirus-mediated transfer of a therapeutic gene, transforming growth factor-β1 (TGF-β1), to the disc was investigated in vivo [18]. Adenovirus constructs (Ad/CMV-hTGFβ1) containing a human TGF-β1 encoding gene with saline was injected into the NP of lumbar discs of skeletally mature female New Zealand white rabbits by anterior approach. Disc NP tissues injected with Ad/CMV-hTGFβ1 showed a 30-fold increase in active TGF-β1 production, and a 5-fold increase in total (active + latent) TGF-β1 production compared to the intact control discs. These tissues also showed a 2-fold increase in proteoglycan synthesis compared to the control discs. This study demonstrated the efficacy and the potential application of adenovirus-mediated gene therapy to disc diseases.
3) Adeno-associated virus
Lattermann et al. [44] reported adeno-associated virus (AAV) vector-mediated gene transfer to the disc. Although adenoviral vectors have high efficacy of transduction in vitro and in vivo, there remain concerns about the immunogenicity and safety in clinical applications, such as fatal anaphylactic reactions after systemic injection [45]. The AAV vectors are potentially less immunogenic than adenoviral vectors and have not been related with any known diseases in humans or mammals [46]. In this study, human disc NP cells were transduced in vitro, and adult female New Zealand white rabbits were injected with AAV vectors carrying different marker genes in vivo. The AAV vectors efficiently transduced human as well as rabbit disc cells and exhibited a robust transgene expression. The overall transgene expression was approximately half of that seen with the adenoviral vectors, and the in vivo gene expression was maintained for 6 weeks. The AAV vectors might offer an alternative to the adenoviral vectors for the treatment of disc degeneration.
4) Baculovirus
Liu et al. [47] reported the utility of baculovirus for the transfer of the green fluorescence protein (GFP) gene to the disc. In vitro, intervertebral disc cells were treated with 6 different doses of baculovirus carrying the GFP gene (Ac-CMV-GFP). In vivo, the Autographa californica nucleopolyhedrovirus/GFP virus was injected directly into discs of New Zealand white rabbits. In vitro, the highest transduction ratio (approximately 87% of disc NP cells) and the long-term expression was found in the dose of Ac-CMV-GFP at a multiplicity of infection of 200 without any toxicity to the cells. The in vivo assay showed that Ac-CMV-GFP could also mediate GFP expression in rabbit disc cells without inducing any symptoms. These results suggested that the baculovirus might be useful tools as a gene therapy vector for disc diseases.
5) Lentivirus
Liu et al. [48] reported in vivo cotransduction of transforming growth factor-β3 (TGF-β3), connective tissue growth factor (CTGF), and TIMP-1 with a lentiviral vector in a New Zealand white rabbit annular puncture model. Lentiviruses can deliver a significant amount of exogenous genes and has an advantage in a multiple gene expression system. This study showed upregulated gene expression and protein synthesis of aggrecan and collagen type II by lentivirus-mediated TGF-β3, CTGF, and TIMP-1 cotransduction. In addition, significant improvement in magnetic resonance imaging (MRI) grading scores was found in the experimental disc compared to the control. These results indicated the feasibility of multiple gene transfer and potential therapeutic approach with lentivirus for the treatment of disc degeneration.
3. Non–Virus-Mediated Gene Therapy
From a safety viewpoint, the problem with retroviral vectors was the risk of insertional mutagenesis while the problem with adenoviral vectors was the immunogenicity of transduced cells [49,50]. In clinical trials, several patients died after viral vector administration [51]. In addition, virus-mediated gene therapy requires exclusive facilities and techniques in manufacturing, specialized patient care units in clinical application, and high costs in all these steps. On the other hand, non–virus-mediated gene therapy has increased attention due to its safety and simplicity [52]. Although various types of non–virus-mediated gene transfer techniques have been investigated, a major limitation has been the lower transfection efficacy compared to viral vector methods.
1) Microbubble-enhanced ultrasound technique
An appropriate intensity of ultrasound exposure has been suggested to make a small transient hole on the cell surface in a phenomenon called sonoporation without cell toxicity, which may increase the permeability of the cell membrane to large molecules such as plasmid DNA [53,54]. Furthermore, Lawrie et al. [55] reported that microbubble echocontrast agents enhanced acoustic cavitation and lead to a distribution of material over a specific area of interest. Both sonoporation and cavitation were considered to have synergistic effects for increased transfection efficiency [55].
Nishida et al. [56] demonstrated this microbubble-enhanced ultrasound gene therapy in the intervertebral disc. Two different reporter plasmid DNA encoding GFP and Firefly luciferase were used. Plasmid DNA was mixed with microbubbles and injected into coccygeal intervertebral discs of Sprague-Dawley (SD) rats. The therapeutic ultrasound was irradiated on the surface of injected discs. Substantial GFP-positive cells were found in the NP from the GFP-transfected group at 1 week. The ultrasound group demonstrated approximately an 11-fold increase in luciferase activity compared to the plasmid DNA-only group. Furthermore, mediated transgene expression was observed at least up to 24 weeks. This study indicated that the microbubble-enhanced ultrasound transfection was a feasible technique for local gene therapy within the disc to enhance the transfection efficacy of plasmid DNA.
2) Polyplex micelle
Another gene delivery method recently applied for the intervertebral disc is the polyplex micelle which forms a corona of hydrophilic segments surrounding the core of water-incompatible segments [57]. Feng et al. [58] reported a sustained and bioresponsive 2-stage microRNA (miRNA) delivery system into disc NP cells with injectable MMP-degradable hydrogels encapsulating MMP-responsive polyplex micelles. In this study, miRNA-29 (miR-29) was focused on. The miR-29 family has the potential to suppress tissue fibrosis, which contains miR-29a, miR-29b, and miR-29c [59]. Cationic block copolymers were designed to complex miR-29a, and subsequently mixed with the polyethylene glycol (PEG) gelation precursors and MMP-cleavable peptide cross-linkers for in situ formation of polyplex micelle-encapsulated hydrogels. In the first stage, elevated MMP levels in the local fibrosis regions triggered the degradation of the hydrogels, thus localizing the sustained release of the polyplex micelles in pathological disc tissues. At the second stage, released polyplex micelles were responsive to MMPs for the detachment of PEG shells, and subsequently, enhanced cellular uptake and endosomal escape can be achieved in disc NP cells. In vitro experiments with rabbit disc NP cells and in vivo analysis with SD rats demonstrated that miR-29a effectively silenced MMP-2 expression, inhibited the fibrosis process, and reversed disc degeneration in animal models through blocking the β-catenin translocation pathway from the cytoplasm to the nucleus, indicating that the two-stage bioresponsive local miRNA delivery system represented a novel strategy for the treatment of chronic disc degeneration.
3) RNA interference
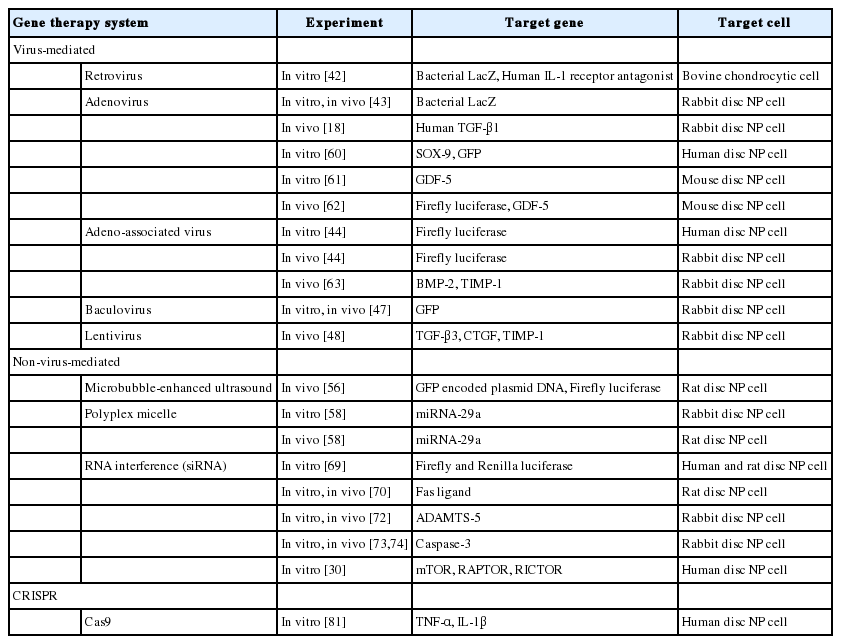
The diversity of signaling molecules in intervertebral disc homeostasis has identified a variety of potential therapeutic genes; e.g., TGF-β1 [18], SRY-box transcription factor 9 [60], growth and differentiation factor-5 [61,62], bone morphogenetic protein-2 [63], and TIMP-1 [63]. These genes have been applied to stimulate or upregulate matrix synthesis, which needs a lot of energy and resources. However, different approaches requiring less energy or fewer resources may be feasible to treat disc diseases under the harsh environment. One of the approaches is the down-regulation of harmful gene expression for the physiological condition of the disc. This strategy focuses more on prophylactic aspects of disc degeneration and theoretically leads to a regenerative treatment over an extended period (Fig. 1).
Schematic illustration of the gene therapy-mediated modification of intervertebral disc cell matrix turnover. Gene therapy either by upregulating matrix synthesis or downregulating harmful gene expression aims the improvement of imbalanced matrix turnover between anabolism and catabolism, which could lead to the deceleration as a prophylactic aspect and hopefully the partial recovery as a regenerative medicine of disc degeneration over an extended period. The transforming growth factor-β1, SRY-box transcription factor 9, growth and differentiation factor-5, bone morphogenetic protein-2, and tissue inhibitors of metalloproteinase-1 are recognized as potential target genes for treatment to upregulate disc matrix synthesis. Then, RNA interference is an applicable therapeutic strategy to downregulate harmful gene expression in the disc.
RNA interference (RNAi) has been developed as an important biological approach for specific gene silencing. The RNAi was first reported in 1998 as the phenomenon observed in Caenorhabditis elegans as a biological response to exogenous double-stranded RNA and a process of sequence-specific, posttranscriptional gene silencing [64]. Elbashir et al. [65] reported gene-specific suppression in mammalian cell lines by 21- or 22-nucleotide small interfering RNAs (siRNA) without stimulating a host interferon response. One of the advantages of siRNA-mediated RNAi is its effectiveness. Even a relatively small amount of siRNA provides effective down-regulation of specific gene expression because siRNA is incorporated into the RNA-induced silencing complex (RISC) that is stable in the cell [66,67] and cleaves repeatedly to the target mRNA [68,69].
Kakutani et al. [69] showed for the first time that the siRNA targeting exogenous reporter gene was effective in silencing the transgene expression in disc NP cells in humans and SD rats in vitro. In this study, 2 reporter luciferase plasmids (Firefly and Renilla) were used. These plasmids were cotransfected with siRNA targeting Firefly luciferase to disc NP cells extracted from rats and human scoliosis patients. The Firefly luciferase expression was significantly inhibited both in rats (94.7%) and in humans (93.7%). These inhibitory effects continued for 3 weeks. This study thus demonstrated that siRNA-mediated gene silencing in rat and human disc cells in vitro is feasible and effective in downregulating specific gene expression.
In the following study, Suzuki et al. [70] reported prolonged effectiveness of the DNA vector-based RNAi technique in vitro. Furthermore, the simple unmodified siRNA-mediated RNAi kept effective in rat coccygeal intervertebral discs for at least 24 weeks. Additionally, this study demonstrated that long-term down-regulation was possible not only for the exogenous reporter gene but also for the endogenous gene (Fas ligand) [71] expression in rat discs in vivo.
As other therapeutic target genes in RNAi, ADAMTS-5 and caspase-3 have been studied. Seki et al. [72] reported an effective suppression of disc NP degradation by a single injection of ADAMTS-5 siRNA in vivo. They confirmed the knockdown rate of ADAMTS-5 siRNA for disc NP cells from Japanese white rabbits in vitro, and conducted in vivo experiment with the New Zealand white rabbit annular puncture model. The ADAMTS-5 siRNA showed significantly improved MRI and histological grading scores compared to the control, although no significant difference was observed in radiographic disc height between the groups during the experimental periods. Sudo et al. [73] and Yamada et al. [74] similarly demonstrated the effects of caspase-3 siRNA in vitro and in vivo. After confirming antiapoptotic effect of caspase-3 siRNA in vitro, they performed in vivo study by using annular puncture model and external compression model respectively. The MRI and histological analysis revealed the suppression of degenerative changes and the inhibition of apoptosis in NP by local injection of caspase-3 siRNA. Although long-term silencing effect of single intradiscal administration of siRNA is not clear enough, RNAi may be suitable as a local gene therapy for disc degeneration and associated disorders by downregulating harmful genes for the normal physiology of the disc.
4. CRISPR-Cas9
The clustered regularly interspaced short palindromic repeats (CRISPR) technology enabled precise and efficient genome editing in living eukaryotic cells. The CRISPR is an adaptive immune system used by microbes to defend themselves against invading viruses by recording and targeting their DNA sequences [75]. Jansen et al. [76] named the repetitive DNA sequences in the prokaryotes CRISPR, which shared common set of features, and identified 4 CRISPR-associated (cas) genes in 2002. Barrangou et al. [77] demonstrated in 2007 that CRISPR and associated cas genes provided resistance against phages, and resistance specificity was determined by spacer-phage sequence similarity. Thereafter, Jinek et al. [78] found that Cas9, the hallmark protein of type II CRISPR-Cas systems, was a DNA endonuclease guided by dual RNAs and showed site-specific double-stranded breaks in target DNA by a Cas9 endonuclease in 2012. Finally, Cong et al. [79] successfully conducted mammalian genome editing with CRISPR-Cas9 system in 2013. In addition, this system has been shown to have minimal off-target effects [80]. Today, CRISPR-Cas9 techniques can be used not only to knockout but also to repair and/or regulate gene expression.
In the field of the intervertebral disc, Farhang et al. [81] reported the receptor-specific inhibition of tumor necrosis factor-α (TNF-α) and IL-1β signaling in human disc cells in vitro. In this study, lentiviral CRISPR epigenome editing systems were introduced into human degenerative disc cells to downregulate TNF receptor 1 (TNFR1)/IL-1 receptor 1 (IL1R1) expression. The transduction of epigenome editing systems was efficient. Although TNFR1 expression was significantly downregulated, IL1R1 down-regulation was not maintained in the majority of the patient samples. Aggrecan increased and MMP-3 decreased in expression significantly by TGFR1 epigenome editing, while aggrecan and MMP-3 did not significantly change in IL1R1 epigenome-edited cells. These results demonstrated the efficacy and feasibility of CRISPR-Cas9 system in pathological disc cells and also revealed a limitation in epigenome targeting of IL1R1, which indicated that a tailored approach may be required for successful regulation of each gene.
AUTOPHAGY AND mTOR SIGNALING
Autophagy is an important cell survival mechanism that sustains metabolism and prevents the accumulation of damaged, toxic proteins and organelles under stress conditions, especially nutrient deprivation [29,32-34]. Autophagy involves autophagy-related (Atg) genes and proteins [32,34]. When cells are under stress conditions, the Atg proteins are activated and form the autophagosome, which captures damaged organelles, misfolded proteins, and invading microorganisms in induced autophagy [32-34]. The completed autophagosome fuses with the lysosome and become the autolysosome, which degrades the enclosed cargo and releases its constituents for reuse [32-34]. The microtubule-associated protein 1 light chain 3 (LC3) (a mammalian homolog of yeast Atg8) is a ubiquitin-like protein with cytosolic (LC3-I) and phosphatidylethanolamine-conjugated (LC3-II) forms [82,83]. The LC3-II is the only protein that keeps the attachment to the autophagosome membrane after processing, thus it is a robust marker for ongoing autophagy [82,83]. The p62/sequestosome 1 (p62/SQSTM1) is a ubiquitin-binding protein that acts as a link between LC3 and ubiquitinated substrates [83]. The p62/SQSTM1 and p62/SQSTM1-bound polyubiquitinated proteins become incorporated into the completed autophagosome and are degraded in the autolysosome; therefore, their expression levels inversely correlate with autophagosome degradation levels [83]. Monitoring this dynamic, sequential process, known as autophagic flux, is essential to understand the roles of autophagy.
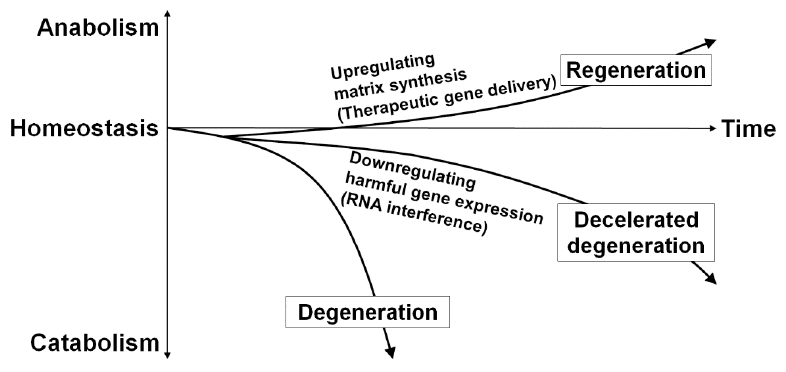
In molecular signaling, autophagy is under the tight negative regulation by the mammalian target of rapamycin (mTOR) [84]. mTOR is a serine/threonine kinase that integrates nutrients, growth factors, energy, and stress signals to trigger the activation of cell growth and division [84]. mTOR exists in 2 complexes: mTOR complex 1 (mTORC1), which contains the regulatory-associated protein of mTOR (RAPTOR), and mTOR complex 2 (mTORC2), which contains the rapamycin-insensitive companion of mTOR (RICTOR) [84]. Downstream mTORC1 effectors, including the p70/ribosomal S6 kinase (p70/S6K), regulate cell proliferation, mRNA translation, and protein synthesis [84], mTORC1 is regulated upstream by Akt, an essential pro-survival mediator that suppresses apoptosis [85]. Moreover, Akt phosphorylation has been associated with the class-I phosphatidylinositol 3-kinase (PI3K) and mTORC2 [84,85]. Because mTOR integrates the signal for nutrition [84], the extensive modulation of mTOR signaling would be harmful, and homozygous mTOR deletion results in embryonic lethality [86]. Therefore, identifying which subunit(s) of mTOR influence on target cells is necessary (Fig. 2).
Schematic illustration of the RNA interference-mediated modulation of intervertebral disc cellular mTOR signaling. The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that detects nutrients to signal the execution of cell growth and division. mTOR exists in 2 complexes: mTOR complex 1 (mTORC1), which contains regulatory-associated protein of mTOR (RAPTOR), and mTOR complex 2 (mTORC2), which contains rapamycin-insensitive companion of mTOR (RICTOR). mTORC1 works as a signal integrator for nutrients, growth factors, energy, stress, DNA damage, and hypoxia. Downstream effectors of mTORC1, including p70/ribosomal S6 kinase (p70/S6K), regulate cell proliferation, messenger RNA (mRNA) translation, and protein synthesis. Autophagy, an intracellular degradation system, is under the tight negative regulation of mTORC1. mTORC1 can be regulated upstream by Akt, an essential pro-survival mediator that suppresses apoptotic cell death. Akt phosphorylation is governed by the class-I PI3K and mTORC2. Furthermore, a negative feedback loop exists between p70/S6K and the class-I PI3K. In our previous study using RNA interference, small interfering RNAs (siRNAs) against mTOR, RAPTOR, and RICTOR were applied to modulate mTOR signaling. In human disc nucleus pulposus cells, RNA interference treatments clarified mTOR-dependent senescent cell aging and extracellular matrix catabolism. The selective suppression of mTORC1/RAPTOR, but not the extensive suppression of mTORC1/mTORC2/mTOR and mTORC2/RCTOR, protected against inflammation-induced disc cellular apoptosis, senescence, and matrix catabolism through the induction of autophagy and Akt.
Little is known regarding the effects of autophagy and mTOR signaling on disc cell and tissue homeostasis. Therefore, the authors performed in vitro studies of human and rabbit disc cells and tissues, modulated by the graded supply of nutrition and RNAi to elucidate the involvement and roles played by autophagy and mTOR signaling in the intervertebral disc.
Yurube et al. [20] conducted time-course rabbit disc AF cell culture in different media with varying serum concentrations under 5% oxygen. Serum and nutrient deprivation decreased disc AF cell proliferation and metabolic turnover and increased autophagy, apoptosis, and senescence. These results demonstrated the serum deprivation induced autophagy in disc cells as shown in prior reports [87-90].
Ito et al. [30] demonstrated the cascade-dependent, differential roles for mTOR signaling in human disc NP cells and the protection against inflammation-induced disc cellular apoptosis, senescence, and matrix catabolism by the selective interference of mTORC1/RAPTOR in vitro. In this study, RNAi of mTOR targeting mTORC1 and mTORC2, RAPTOR targeting mTORC1, or RICTOR targeting mTORC2 was used. RNAi of mTOR and RICTOR decreased p70/S6K and Akt phosphorylation, while RAPTOR RNAi decreased p70/S6K but increased Akt phosphorylation. All RNAi treatments increased LC3-II and decreased p62/SQSTM1, indicating enhanced autophagy. IL-1β-induced apoptosis decreased by RAPTOR RNAi. IL-1β-induced senescence-associated β-galactosidase (SA-β-gal)-positive cells and p16/INK4A expression also decreased by RAPTOR RNAi. In matrix metabolism, RAPTOR RNAi reduced IL-1β-induced catabolic MMP and upregulated anticatabolic TIMP expression. Selective interference of mTORC1/RAPTOR protected against inflammation-induced apoptosis, senescence, and matrix catabolism possibly through Akt and autophagy induction in human disc cells.
CONCLUSION
The difficulty in disc regeneration using biological approaches originates from the unique anatomical and physiological character of the disc. Under the limited nutrition and oxygen especially in the central NP region, the disc is not able to support abundant cell survival, especially consuming energy. The RNAi is one of the possible solutions and has been applied to downregulate catabolism in the disc and correct imbalance between anabolism and catabolism with minimal nutrition requirements. Although this kind of approach focuses more on the prophylactic treatment of disc degeneration, it could have the potential as a regenerative treatment over an extended period. The authors found the in vivo involvement of mTOR signaling in human degenerative disc NP tissues from patients. Therefore, we propose gene therapy, targeting mTORC1/RAPTOR disruption as future treatment strategies for degenerative disc disease. There are many obstacles to be overcome in disc regeneration research, such as safety aspects, high costs, and transfection efficacy, before findings can be clinically applied. However, the amount of research is increasing and broadening, and so it is not unrealistic to expect a breakthrough from these studies over the coming few years.
Notes
The authors have nothing to disclose.