






INTRODUCTION
Spinal cord injury (SCI) is the destructive neurological damage to the spinal cord that can cause temporary or permanent sensory and motor impairment in patients affecting their quality of life [1,2]. Injury to the spinal cord may be of exogenous or endogenous origin, of which more than 90% of cases are considered traumatic in etiology and affect more adult males as compared to females [2]. Traumatic causes of the injury may include sports or vehicular accidents, falls, bullet injuries, or any other forms of violence, whereas the nontraumatic injury may be due to acute or chronic illnesses, including tumors and infections.1 Depending upon the time sequence of the injury, SCI’s overall pathophysiology is split into 3 major phases: acute, subacute, and chronic, with each phase presenting progressive damage to the spinal cord over time [3]. At the initial stages, the traumatic injury (primary injury) only affects the spinal cord mechanically by damaging the blood vessels and cellular membranes, causing ischemia and inflammation. This is followed by the secondary injury, a cascade of events that further exacerbates the neurological damage through vascular, biochemical, and cellular changes. Most of the secondary events (subacute and chronic phase) in SCI pathophysiology include complex mechanisms leading to calcium and glutamate excitotoxicity, vascular changes, ionic imbalance, reactive oxygen species (ROS) production, lipid peroxidation, inflammation, and apoptosis. This complex secondary cascade at later stages initiates the development of a glial scar and cystic cavities that impairs the normal myelination and axonal regeneration process resulting in a permanent detrimental state of the injury [1,3].
Glutamate excitotoxicity during the postinjury period is one of the most concerning states responsible for connecting several other mechanisms, furthering the damage. Several studies report the role of overactivated N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors in raising sodium and calcium levels intracellularly. In addition to the toxicity in the myelinated axonal region, the overactivation of the glutamate receptor, AMPA, in the white matter region is toxic to oligodendrocytes and astrocytes [4]. The glutamate receptor overactivation and the downregulation of glutamate transporters are the leading causes of persistent hyperexcitability [5]. Initially, acute primary damage to the spinal cord gives rise to hemorrhage at the injury site. Further, this results in ischemic insult due to ATP depletion at the site, raising energy demands, thus reducing the normal transportermediated uptake of glutamate [2]. Downregulation of astrocytic GLT-1 (glutamate transporter 1) and GLAST (glutamate/aspartate transporter) postinjury period has already been reported [6]. Besides this, injury-associated blood-brain barrier (BBB) disruption releases excitatory amino acids (EAAs) through nonspecific membrane micropores leading to astrocytic edema and glutamate extravasation. Altogether, these factors, receptor overactivation, downregulation of transporters, and BBB disruption contribute to raising the glutamate levels extracellularly. Furthermore, glutamate-mediated sustained depolarization causes heavy calcium influx through NMDA, AMPA, and G protein-coupled glutamate receptors, resulting in an ionic imbalance that increases the lesion damage, neuronal and glial cell loss in the spinal cord [4,6].
Persistent glutamate-mediated hyperexcitability contributes to the insufficiency of GABAergic inhibitory tone in the spinal cord dorsal horn (DH), which is reported to be one of the leading bases for the emergence of chronic neuropathic pain (CNP) post-SCI [7]. Previously Zhang et al. [8] reported the loss of GABAergic cells in the lumbar region after photochemically induced spinal cord ischemia in rats. Another study showed that an early rise in oxygen free radicals due to oxidative stress caused a reduction in GABAergic neurotransmission by modulating GABAA gated chloride channels [9]. Also, alterations in levels of cation-chloride channels (upregulation of Na+-K+-Cl- cotransporter isoform 1 [NKCC1] and downregulation of K+-Cl- cotransporter isoform 2 [KCC2]) was observed after the injury on which GABAergic tone is dependent [10]. There was also a loss of the enzyme glutamic acid decarboxylase (GAD) isoform GAD65 noticed in spinal DH post nerve injury, which is responsible for synthesizing GABA [11]. Gene deletion of synapsin II by inhibiting glutamate release from primary afferent nociceptive fibers and promoting long-lasting GABA release alleviated mechanical and cold allodynia symptoms in neuropathic pain (NP) mouse model [12]. Together, these changes outline the insufficiency of inhibitory GABAergic tone after the central nervous system (CNS) injury. Inhibiting this tone using GABA antagonists has shown NP like effects [13]. Hence, raising GABAergic tone post-SCI remains a significant approach for alleviating CNP.
Another major complication of chronic nature after the injury is spasticity. It is one of the components of upper motor neuron (UMN) syndrome with hypertonia, hyperreflexia, and clonus-like symptoms. Various reports provide evidence of hypoactive GABAergic inhibition post-SCI as a major cause of spasticity development [14]. Thus, enhancing GABAergic tone for easing the spastic symptoms is already a promising intervention that suggests the role of hypoactive GABA in causing spasticity. Several mechanisms of a complex nature have led to the development of spastic symptoms; hence absolute treatment to relieve spasticity remains challenging after the injury [14]. In this review, we explore the role of GABA in producing CNP and spasticity which is experienced by most of the patients in post-SCI period. Several approaches that specifically target the inhibitory GABAergic tone for attenuating both these complications will be discussed in detail with special emphasis on ongoing research.
GABA
1. Synthesis, Release and Spinal Distribution
Even though glutamate is an excitatory neurotransmitter, it synthesizes GABA, which is the primary inhibitory neurotransmitter in the brain. Glutamate is the standard source for the synthesis of GABA in CNS using the enzyme GAD. In humans, GAD exists in 2 isoforms, GAD65 and GAD67. Both the isoforms are expressed differently in brain regions. It is considered that 90% of GABA is produced utilizing the GAD67 isoform. Besides glutamate, GABA can also be synthesized using polyamine, ornithine, arginine, or homocarnosine pathways through putrescine using various enzymes [15].
Conventionally, GABA is released from the axonal terminals of neurons by calcium-mediated vesicular exocytosis [16]. However, a few reports suggest the release of GABA by some unconventional means. Dendritic release of GABA in a retrograde manner through high depolarization mediated release of glutamate in the olfactory bulb and astrocytic GABA release through astrocytic synthesis or uptake mechanisms are the examples for unconventional GABA release [16].
GABA is abundant in the spinal cord, it is presented by interneurons only and not by the projection neurons [17]. GABAergic interneurons are localized largely in superficial laminae, while some are in deeper laminae of the dorsal and ventral horn. The inhibitory part of the GABAergic system is also distributed through various synaptic organizations that are axosomatic or axoaxonic. Besides this, GABAergic myelinated fibers found in the white matter region also indicate the inhibitory system’s presence in the supraspinal region [18].
2. GABA Action – GABA-A, B, and C Receptors
The GABAergic inhibitory system functions by acting on 3 main receptors, GABAA, GABAB, and GABAC, which are present on pre or postsynaptic membranes of neurons and glial cells [19]. Structurally, GABAA is a pentameric ligand-gated inotropic receptor complex having subunits with various isoforms. The binding of GABA to this receptor in the unoccupied state is quite high, which causes a conformational change leading to postsynaptic Cl- channel pore opening and thus allowing Cl- influx-mediated hyperpolarization. Several exogenous compounds like benzodiazepines, barbiturates, picrotoxin possess binding sites on the GABAA receptor, hence act as modulators [15].
GABAB receptors are metabotropic G protein-coupled receptors linked with Ca2+ and K+ channels. They primarily have 2 receptor subunits GABAB R1 and GABAB R2. These receptors are localized either in the presynaptic region as autoreceptors that regulate the GABA release or in the postsynaptic region that causes hyperpolarization by activating K+ channels [15].
GABAC receptors were unresponsive to both bicuculline (GABAA antagonist) and baclofen (GABAB agonist). This gained attention as it illustrates, they are distinct from the GABAA and GABAB receptors. Though their inotropic and ρ subunits (ρ1–ρ3) containing properties are identical to GABAA receptors, they differ in that they are approximately 10 times more sensitive to GABA, have a greater number of binding sites, possess a poor ability to undergo desensitization, have homooligomeric assembly of ρ subunits (ρ1–ρ3) and prominent localization to retina rather than the entire CNS [15,19].
3. Glutamate-Glutamine-GABA Cycle Homeostasis
The glutamate-glutamine-GABA cycle helps balance the amino acid neurotransmitters GABA and glutamate, employing neuronal and glial transfer. In glutamatergic neurons, glutamate is synthesized using the tricarboxylic acid cycle (TCA) intermediate α-ketoglutarate. This is then taken up by astrocytes and is changed into glutamine using the enzyme glutamine synthetase. After astrocytic release, the extracellular glutamine is taken up by GABAergic neurons. Glutamate is produced using the enzyme glutaminase, which ultimately produces GABA using the GAD enzyme [20]. Similarly, uptake of released GABA by astrocytes converts GABA to TCA intermediate succinate by the enzyme GABA transaminase and succinate semialdehyde dehydrogenase. This succinate can synthesize glutamine and further α-ketoglutarate via citrate. This makes way again for α-ketoglutarate to synthesize glutamate and the cycle continues [21].
GABA AND NEUROPATHIC PAIN POST-SCI
Quality of life post-SCI is often quite poor. Furthermore, centrally initiated NP observed in two-thirds of the injured patients for chronic periods adds up more to the same [22]. The pain of conventional inflammatory causes is protective, which generally subsides with the healing process of the injury or recovery from the disease. However, NP brought about by injury or disease of CNS, can last even after the resolution of disease or injury healing without giving any protective assistance to the patient [23]. International Association for the Study of Pain classified CNP of SCI based on the location of its emerging, that is either above-level, at-level, or below the level of injury which may last for months or throughout the lifetime of the patients. Patients generally experience CNP differentially. Some describe it to be continuous, some interrupted, while in some, it is spontaneous or stimulus-induced [24]. At-level pain generally involves allodynia, spontaneous and consistent pain representing the retention of partial sensory activity at the site [22]. Factors like the age of more than 50 years, tetraplegia, and the traumatic cause of the injury make SCI patients more at risk of CNP. Most of the patients develop below-level pain after 6 months to one year of the injury [25]. Several cellular, biochemical or electrophysiological mechanisms are responsible for CNP, of which neuronal and glial hyperexcitability, microglial activation, and alterations in somatosensory cortical reorganization majorly contribute. To study causal mechanisms of CNP, various animal models have been made, including inducing ischemic lesions, anterolateral spinal cord lesions, transection, clip compression, contusion, hemisection, quisqualic acid injection resulting in mechanical and thermal hyperalgesia and allodynia. The contusion model is more frequently used to study NP-based mechanical hypersensitivity [22,26].
The spinal cord plays a central role in nociceptive and nonnociceptive signal processing and control through afferent pathways carrying further inputs to the brain. Nociceptive neurons in the DH of the spinal cord having terminals of nociceptive afferent fibers Aδ and C fibers, project signals to the brain by direct and indirect inputs, respectively. The neurons in lamina II of the DH are either excitatory or inhibitory and show a response mostly to the received nociceptive inputs [27]. Normally, there is a counterbalance between the excitatory and the inhibitory inputs. The inhibitory interneurons of the spinal cord limit the spinal nociception by pre and postsynaptic inhibition mechanisms mediated by GABAergic synapses and GABA and glycine synapses, respectively. The direct antinociceptive descending inhibition through GABA and glycine pathways also help in controlling nociceptive inputs, which are activated by bulbospinal projections of the brain stem [17].
In contrast to this, some reports suggest that inhibitory GABAergic transmission may transmit pain impulses rather than inhibiting them. Nevertheless, the activation of GABAA and GABAB receptors, in general, have shown antinociceptive effects [28]. The presence of lesions or injury in the nervous system is responsible for CNP. The excitotoxic damage leads to the release of excitatory glutamate from afferent fibers. Further, glutamate in excess over activates its receptors causing high calcium influx, increasing persistent hyperexcitability and central sensitization of DH neurons leading to NP [27]. The release of neuropeptides, ROS, adenosine monophosphate, and inflammatory cytokines due to neuronal and glial cell’s hyperexcitability further enhances pain transmission [29]. Sensory neurons experience phenotypic changes after the injury. Neurons are reported to possess more of wide dynamic range (WDR) neuron properties making them respond to both weak as well as strong stimuli in the DH [29]. Alterations in pathways of sensory neurons allow painful response to nonnoxious stimuli as the noxious, which is referred to as allodynia, generally experienced in NP [29]. Many such mechanisms are responsible for an imbalance between inhibitory and excitatory tone in the postinjury period that is discussed later in this review.
SCI DIFFERENTIALLY REGULATES GABA LEVELS
The complex pathophysiology of SCI involves various events promoting imbalance between the excitatory-inhibitory tone by altering the levels of amino acids released in the DH. Panter et al. [30] described that an early rise in levels of these amino acids were linked with the degree of traumatic injury induced in rabbits. The more severe the injury, the greater the increase in the levels of all 9 amino acids along with GABA and glutamate were observed. Similarly, in rats, the acute traumatic contusive injury produced an initial rise in GABA levels at early hours postinjury [31]. This initial upsurge was observed due to increased GAD levels, which synthesized more GABA in the early 12 hours postinjury [32]. Initial damage to the GABAergic cell membranes, more release of GABA from synaptic terminals, GABA uptake transporter downregulation are considered to be the primary reasons for the early rise in GABA levels after injury [33].
In contrast to the above findings, the overall inadequacy of the inhibitory GABAergic tone promoting CNP is already well established [8,34]. Persistent hyperexcitability in the postinjury period causes the overactivation of glutamatergic neurons, promoting cell death [6]. Also, Fas/CD95, tumor necrosis factor R1 (TNFR1), and TNFR2 signaling mediated cell death of neurons and glia in the spinal cord is reported after the injury, which could relate to the loss of GABAergic interneurons [35]. Meisner et al. [34] reported the GABAergic neuronal loss in the DH post contusive injury, which led to mechanical and thermal hyperalgesia in mice, that was then relieved by tiagabine, an inhibitor of GABA transporter. The expression of protein GAD65, which is responsible for synthesizing GABA from glutamate, was downregulated in the DH after the injury suggesting low GABAergic tone [36]. Likewise, reduction in levels of GAD67 was observed in chronic constriction of the infraorbital nerve model, causing mechanical allodynia, which was later attenuated using GABA transaminase inhibitor, vigabatrin [37]. Gosselin et al. [38] reported astrocytic and microglial activation mediated increase in GABA uptake by upregulation of GABA transporter 1 (GAT-1) in the gracile nucleus of spared nerve injury NP models of rats. In one study, administration of GABAA receptor antagonist bicuculline triggered allodynia-like behavior in rats signifying the decreased GABAergic tone, relating it to NP postinjury [39]. Altogether, these studies of GABAergic cell loss, low expression of GAD isoform, and high transporter-mediated GABA uptake provide a sound basis for associating GABAergic tone loss with CNP after the injury.
MECHANISMS REDUCING THE GABAERGIC TONE
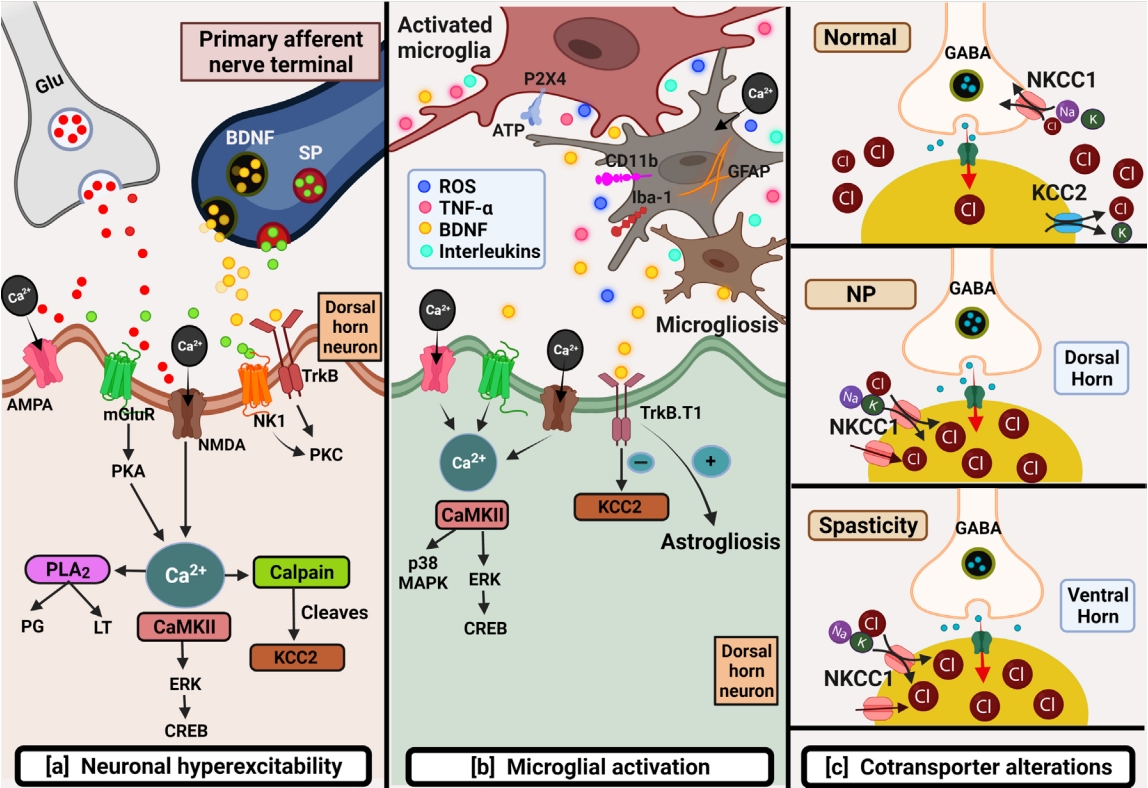
Along with the explanations mentioned above of GABAergic cell loss, high GABA reuptake, and low GAD65/67 proteins, a significant role is played by persistent hyperexcitability, microglial activation, and cation-chloride channel alterations, which together have a role in lowering the GABAergic tone. Also, these mechanisms go hand in hand postinjury to cause CNP (Fig. 1). A study reports that a GABAB agonist baclofen and NMDA antagonist ketamine when given in combination, were found to be more efficacious and worked synergistically to attenuate CNP (Table1) [40]. However, as the definitive treatment to deliver complete relief from NP for long periods remains lacking, many researchers are working on addressing the underlying complex mechanisms to target CNP in SCI patients.
1. Persistent Hyperexcitability Post-SCI
The neuronal hyperactivity is considered the major mechanism in inducing CNP post-SCI [29]. The early rise in toxic levels of EAAs after injury to the spinal cord is already recognized. The rising levels themselves present a threat to healthy cells because of damage due to the overactivation of glutamate receptors. So how do EAAs that are released in just minutes post-SCI, affect the neurons? This was studied by administering exogenous glutamate and aspartate mixture in vivo, which resulted in the loss of neurons due to the excitotoxic secondary damage. Several such studies confirm the neurotoxic nature of EAAs after the injury [41]. Injury to the spinal cord using AMPA agonist quisqualic acid also caused excitotoxic damage resulting in neuronal loss leading to spontaneous and evoked pain responses in rats [42]. Normally, the descending inputs and presence of the inhibitory tone counterbalance the hyperexcitability but after SCI, the loss of inhibitory GABAergic inputs led to disinhibition facilitated neuronal sensitization [43].
Many previous studies link neuronal hyperexcitability in increasing nociceptive transmission. Overactivation of glutamate receptors causes heavy calcium influx, which further initiates several downstream signaling pathways by activating kinases [44]. Upregulation in phosphorylated calcium/calmodulin-dependent kinase II was observed in neurons and oligodendrocytes after SCI contusion in rats. This resulted in WDR neuronal hyperexcitability mediated mechanical allodynia [45]. Heavy calcium influx can trigger phospholipase A2 activation, which in turn can produce prostaglandins and leukotrienes or be a prevailing mediator for the production of ROS, reactive nitrogen specie, and other pathways. ROS through transient receptor potential vanilloid 1 and ankyrin 1 channels can trigger glutamate release and the production of inflammatory cytokines [29]. Lowering the neuronal activation threshold contributes to neuronal hyperexcitability mediated central sensitization [46]. Neuronal hyperactivity is well characterized in SCI-NP animal models signifying electrophysiological changes brought about by long neuronal after discharges, reduced action potential generation thresholds, and alterations in their frequencies [29]. Various studies that target CNP using glutamate receptor antagonists provide more relevance to this. In 1997, it was reported that NMDA antagonist MK-801 and AMPA antagonist NBQX could prevent excitotoxic neuronal loss and provide neuroprotection to the injured spinal cord [47]. Similarly, when these compounds were tested on different phenotypes of lumbar spinal cord neurons of the thoracic SCI hemisection model, they helped in reducing the neuronal hyperexcitability mediated below-level pain by blocking inotropic glutamate receptors [46]. Then Bennett et al. [48], with the use of D-AP5 (NMDA antagonist) and NBQX (AMPA antagonist) demonstrated that inhibiting glutamate overactivation could alleviate mechanical allodynia in SCI rats. Hyperexcitability of neurons after SCI is not just limited to the DH of the spinal cord, rather this inevitable event occurring in the supraspinal region and higher brain centers also has contributed to producing ongoing pain [43].
2. Reactive Microglia and CNP Association
Similarly, various receptors, transporters, ion channels, or signaling pathways presented by neurons are also expressed in glial cells. The mechanical damage to the spinal cord, through varied outcomes, initiates activation of resting microglia, contributing to CNP. Major mechanisms for microglial activation include glutamate-mediated hyperexcitability, stimulated excessive nociceptive neuronal firing in the DH, ionic imbalance, cytokines and chemokines release, T cells and leukocyte infiltration, and alterations in proteins that are regulating the cell cycle. In response to the injury-induced damage to the nerves, it is reported that upon activation, microglia undergo several morphological changes and express various markers like CD11b, glial fibrillary acidic protein, and Iba-1. An increase in CD11b expression was observed from 2 hours and up to 180 days after the injury [49]. In addition, the expression of one such molecule, P2X4 (purinergic receptor), is found to be upregulated on reactive microglia after peripheral nerve injury, which is known to be associated with increased pain hypersensitivity [23].
Reactive microglia by activating signaling cascades like p38 and extracellular regulated kinase mitogen-activated protein kinase (MAPK) pathway, or the cAMP response element-binding protein signaling pathway leads to proliferation and recruitment at the injured site [23,45]. A study conducted by Crown et al. [50] suggests that high expression of neuronal, astrocytic and microglial p38 MAPK resulted in development of mechanical allodynia in rats post-SCI. Activation of transcriptional factors alters target gene expression, which further encourages phosphorylation of ion channels and receptors, upholding neuronal hyperexcitability [29]. These reactive microglia are found not only in the entire spinal axis but also in supraspinal regions like the hippocampus, anterior cingulate cortex, and posterolateral nucleus of the thalamus [49]. Reactive microglia promote regeneration and sprouting of primary afferent fibers by releasing nerve growth factors. They induce maladaptive synaptic reorganization and alter neuronal networks through neuronal-glial interactions. Further, these alterations enable the release of pain mediating substances such as ROS, TNF-α, brain derived neurotrophic factor (BDNF), and interleukins from activated microglia [29]. The BDNF receptor truncated isoform tropomyosin-related receptor kinase type B (TrkB.T1), which is solely present on astrocytes, is upregulated after SCI. This upregulation promotes astrogliosis, further contributing to NP. Deleting TrkB.T1 in knock-out mice decreased the proliferation and migration of astrocytes further attenuating NP [51]. Detloff et al. [52] recognized and correlated the onset of microglial activation with induction of allodynia-like behavior after SCI in rats. Intrathecal administration of propentofylline not only modulated microglial activation but also decreased the downregulation of GAD65. This links microglial activation with reducing inhibitory GABAergic tone. Also, agents such as minocycline, lipoxin A4, resolvin E1, estrogen, and rapamycin, by inhibiting microglial activation could positively attenuate the injury related CNP behavior which signifies the role of microglial activation in CNP [36].
3. Cation-Chloride Cotransporters and CNP Association
GABAergic inhibitory tone in the DH is maintained typically through GABA release from interneurons that can block the nociceptive primary afferent inputs by maintaining chloride concentration. The 2 main cation-chloride cotransporters, NKCC1 and KCC2, support in maintaining chloride homeostasis in the spinal cord [53]. GABAA responses in the initial developmental stage of neurons are considered excitatory, however after birth they develop and shift to produce inhibitory responses due to enhanced KCC2 expression, responsible for causing chloride extrusion. Normally, the expression of KCC2 is specifically high in neurons, whereas NKCC1 is more expressed in the respiratory system and kidneys of mice [54]. Alterations in levels of both the cotransporters are reported to contribute to developing CNP postinjury. Upregulation in the expression of NKCC1 and downregulation in the expression of KCC2 was reported 2–14 days after the injury [10]. Loss of KCC2 causes chloride accumulation thus reducing inhibition required for maintaining chloride homeostasis upon activation of GABAA receptors [22]. Factors like microglial activation and raised BDNF significantly contribute to KCC2 downregulation post-SCI [22]. Recently, in an open label trial, it significantly reduced the pain intensity in SCI patients by enhancing KCC2, confirming its significance in GABAergic disinhibition mediated NP induction (Table 2) [55].
Some studies have emphasized the effect of NKCC1 inhibition in reducing the depolarizing GABAA currents; however, its wide distribution in kidney and respiratory system showing undesirable side effects limit its use [54]. NKCC1 antagonist bumetanide, positively aided in reducing the injury initiated CNP by inhibiting the upregulated transporter. But, some patients reported adverse effects like diuresis, difficulty in evacuation and incontinence [55]. Considering this and low levels of KCC2 (chloride extruder protein) in NP, promoting intracellular chloride efflux by enhancing KCC2 cotransporter or decreasing its downregulation is a promising therapeutic approach for reducing NP. A KCC2 agonist, CLP290, was found to be effective in attenuating peripheral nerve injury pain in rats. It provided analgesia without compromising the motor function [56]. Another study helped promote functional recovery in a double hemisection SCI model of mice with staggered lesions (Table 1) [57]. Further studies with this compound would provide more significance for its use in the future.
RESTORING THE INHIBITORY TONE RELIEVES SCI-CNP
Years ago, the ‘gate control theory of pain’ explained that the spinal cord DH's inhibitory neurons can control the peripherally sensed nociceptive inputs by directing them towards the higher centers [58]. GABAergic drugs have been long used as sedatives, anxiolytics, and antiepileptics. Nevertheless, their usage for relieving CNP is new and needs additional exploration. Anticonvulsant drugs like lamotrigine, topiramate, and gabapentin have been shown to increase GABA levels for 4 weeks in healthy adults [59]. This suggests they might be used during the chronic period in NP.
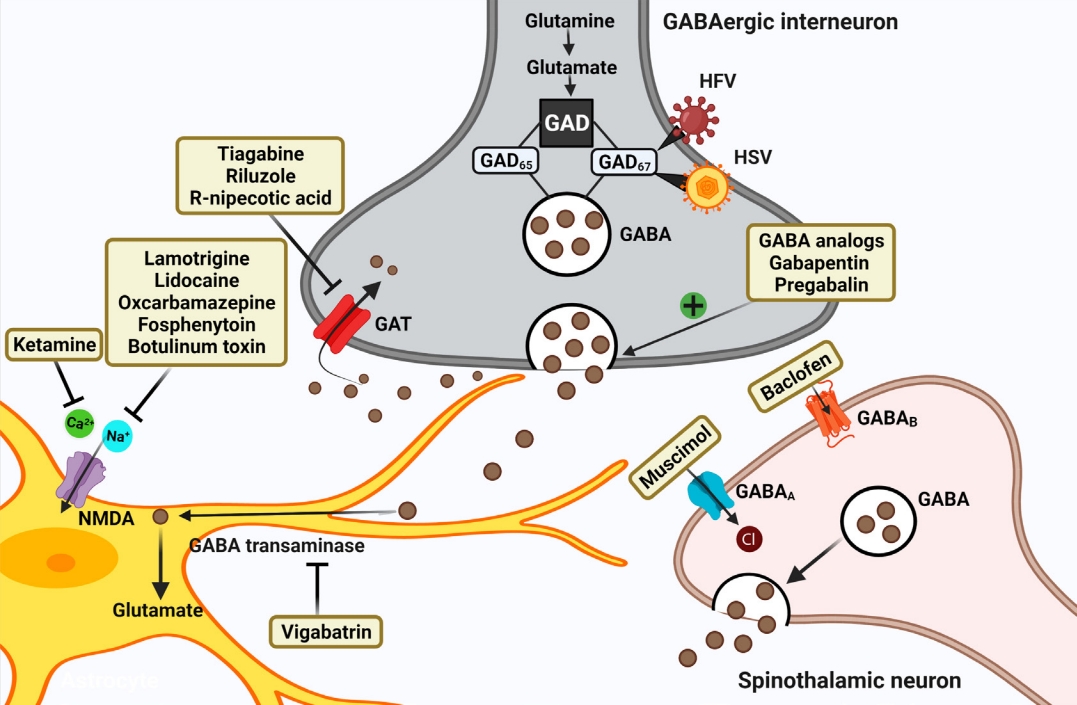
Promoting presynaptic inhibition allows GABAergic neurons to connect with primary afferent Ia fibers, which inhibits EAA release from Ia fibers, further preventing the nociceptive transmission to motor neurons [60]. Several strategies help restore the hypoactive GABAergic tone by acting through diverse mechanisms and hence providing a more fundamental understanding to target GABA-associated CNP (Fig. 2; Tables 1, 2).
1. Enhancing GAD Expression
The approach of enhancing GAD expression to reduce CNP using gene therapy has shown positive results in studies [61]. GAD67 protein can be overexpressed using human foamy virus transduction in rat neurons, resulting in decreased NP in rats (Table 1) [61]. When a herpes simplex virus vector encoding GAD67 was constructed and transduced in the SCI hemisection model, it decreased mechanical allodynia and thermal hyperalgesia by increasing GABA release (Table 1) [62]. Similarly, when a recombinant helper-dependent adenovirus as a vector targeting dorsal root ganglion and overexpressing GAD67 was made, it alleviated allodynia in a spinal nerve transection mice NP model by suppressing Cav3.2 mRNA expression [63]. In another study, exercise training relieved NP-linked mechanical and thermal hyperalgesia, which increased GAD65/67 expression through BDNF elevation. Overall, this suggests that raising GAD levels which are depressed in the postinjury period, could cause more synthesis of GABA to increase inhibitory GABAergic tone. Delivering GABA directly to the site is effective but its short half-life requiring continuous infusion and systemic side effects limits its use. Gene therapy circumventing such drawbacks provides a promising approach for NP attenuation [63].
2. Reducing Hyperexcitability
In clinical studies, various established drugs are being studied to reduce the hyperexcitability and promote inhibitory tone for relieving CNP post-SCI. NMDA antagonist ketamine prevents hyperexcitability mediated central sensitization, thus preventing NP. However, patients in trials experienced disturbing side effects such as sedation, dizziness, and visual distortions limiting its use in SCI [64]. In another trial, when ketamine was given as an adjuvant to gabapentin in low doses for 7 days, it was found efficacious for 2 weeks with only mild side effects (Table 2) [65]. Drugs like lamotrigine, lidocaine, oxcarbazepine, and fosphenytoin block sodium channels further by inhibiting the release of EAA, reducing neuronal firing, and thus are found to be effective in reducing NP post-SCI.
The effect of lidocaine is examined in post-SCI NP topically, intrathecal as well as intravenously [64]. A randomized controlled trial reported that lidocaine relieved at-level and below-level NP when administered intravenous in SCI patients (Table 2) [66]. Another study examined the analgesic effects of sodium amobarbital vs lidocaine. Sodium amobarbital relieved NP post-SCI better than lidocaine by enhancing GABAA inhibition and antagonizing the effects of AMPA, NMDA and kainate receptors (Table 2) [67]. Lidocaine intravenous if given alone showed toxicity when used for a long term [64]. But as a transforaminal epidural injection with depo medrol it resolved pain in patients for months [68]. In one study, lidocaine 5% medicated patch when applied topically reduced CNP after decompression surgery post-SCI (Table 2) [69]. A recent study reports lidocaine as neuroprotective by showing anti-inflammatory and neuron sparing effects in an induced excitotoxicity model without any motor and sensory impairment [70].
Lamotrigine also inhibits the sodium influx-mediated glutamate release, thus reducing neuronal hyperexcitability. In a randomized placebo-controlled trial, it reduced the below-level pain only in a subgroup of patients with incomplete SCI (Table 2) [71]. A Cochrane review published in 2013 suggests no or very less significant evidence showing prominent effects of lamotrigine in NP [72]. However, in a comparative trial 3-week study of amitriptyline versus lamotrigine done in SCI NP patients, both the drugs were found efficacious with no difference in efficacy between them (Table 2) [73].
Botulinum toxin type A (BTX), which is considered as a thirdline treatment for NP, inhibits pain progression by suppressing the release of neurotransmitters and neuropeptides like glutamate, substance P, and calcitonin gene-related peptide [74]. It is also known to reduce spasticity-related post-SCI pain. It relieves mechanical allodynia and hyperalgesia by blocking neuronal sodium channels. Results from a randomized, double-blind placebo-controlled trial suggested that BTX decreased post-SCI pain intensity in patients at weeks 4 and 8 (Table 2) [75]. A recent study reports that BTX reduces the expression of upregulated CXCL13 and GAT-1 in NP chronic constriction injury (CCI) rat models [76]. Presently, lack of studies, low patient numbers, and lack of standardized optimum dose and delivery are the shortcomings that need sound research for BTX approval in NP post-SCI [74].
Already an approved drug for amyotrophic lateral sclerosis, riluzole reduced below-level cutaneous hypersensitivity in SCI-NP rats by inhibiting the glutamatergic transmission (Table 1) [77]. It induced long-term depression from pain fibers to spinal superficial dorsal horn (SDH) neurons, thus inhibiting the excitatory synaptic transmission [78]. Previous reports also suggest that riluzole potentiates GABAergic responses inhibiting the GABA uptake in a dose-dependent manner [79]. A study revealed that inducing the outward current in substantia gelatinosa neurons potentiated the GABAergic transmission in the SDH thus attenuating mechanical allodynia in the NP rat model [80]. Newly synthesized N-alkylated derivatives of riluzole also increased the hot plate latency time and showed antinociceptive effects. Such derivatives carry the potential to be investigated further in NP models [81].
3. Structural Analogs of GABA
Structural analogs of GABA, gabapentin, and pregabalin are the first-line treatment available for NP post-SCI. Their efficacy and tolerability are demonstrated in phase 3 randomized trials of SCI-NP patients (Table 2) [82]. They are known to act by binding α2δ calcium channel subtype, thus reducing spinal excitatory glutamate and substance P release, without showing any serious side effects [26,83]. Other favorable mechanisms include promoting the descending inhibitory pathway, a decrease in glial activation and proinflammatory cytokine release. They also promote axonal regeneration and are found to be neuroprotective in several animal models of neurotrauma. When administered in the acute phase of the injury, they carry the potential to prevent the CNP development [84]. A recent meta-analysis report suggested that compared to the other drugs used, pregabalin was found to be more effective, and gabapentin safer for CNP post-SCI [83]. Gait instability, sedation, and dizziness are some of their common reported adverse effects. Gabapentin may cause cognitive dysfunction. They generally have less tolerance capability as compared to other used drugs like opioids [85].
4. Inhibiting GABA Uptake and Metabolism
Vigabatrin, the irreversible inhibitor of the GABA transaminase enzyme, produced analgesia in the rat NP model [86]. It has also shown neuroprotective effects in spinal ischemia [87]. Tiagabine, riluzole, and R‐nipecotic acid by inhibiting GABA transporters (GATs) prevent GABA reuptake, and increase GABA in order to lessen NP after the injury (Table 1) [28,34,77]. Pyridoxal 5-phosphate serves as a cofactor in GABA reuptake using GATs. Derivatives of hydrazine inhibit the binding of this cofactor and thus reduce the activity of GATs to raise GABA [17].
Although tiagabine is the only clinically approved GAT inhibitor for NP, several molecules and drugs are under investigation for the same [17,88]. In a recent study, novel functionalized amino acids that were designed and synthesized exhibited inhibitory effects on mGAT4 and mGAT2 transporters and showed analgesic effects in preclinical studies of 3 different NP models [88]. Similarly, in another report, novel compounds inhibiting mouse GABA transporters (mGAT3/4), showed an antiallodynic effect by making GABA more available in diabetic and oxaliplatin-induced NP models [89]. Also, GAT-1 inhibitor NNC711 and GAT-3 inhibitor SNAP-5114, by decreasing the amplitude of pain fibers mediated excitatory postsynaptic currents produced analgesia in NP mice models. Further, administration of GABAB antagonist CGP55845 antagonized the analgesic effects suggesting GAT inhibition’s role in producing GABAergic analgesia [90]. These studies could make way for testing these compounds for CNP post-SCI in the near future.
5. GABA Agonists
Muscimol is a GABAA agonist, and baclofen, which is a GABAB agonist, both can attenuate NP by activating the GABAergic receptors to release GABA [91]. Knabl et al. [58] in their work reported that drugs modulating α2 and α3 subunits of GABAA receptors and sparing α1 subunit would be more antinociceptive for relieving chronic pain. α2 GABAA receptor modulation and maintaining chloride homeostasis using KCC2 enhancer when combined worked synergistically to attenuate NP post nerve injury [92]. Muscimol by stimulating GABAA receptors has alleviated NP symptoms in various animal studies of nerve injury models [93]. A recent study reported that muscimol, by reducing WDR neuronal overactivity suppressed both mechanical allodynia and thermal hyperalgesia in CCI-induced rats [94]. The combination therapy of muscimol and endomorphin-1 intrathecal raised the pain threshold in the SCI-NP rat model. This study also reported an increase in the expression of α2 GABAA receptor subunit post-administration (Table 1) [95].
Baclofen is already used for spasticity. It showed an anti-allodynic effect in rats with induced spinal ischemia, reversing WDR neuronal hyperexcitability [96]. Several other preclinical findings report its antinociceptive property in NP by promoting GABAergic tone. In a case report, intrathecal baclofen combined with clonidine relieved intractable post-SCI NP [97]. In a randomized, double-blind, placebo-controlled trial, intrathecal administered baclofen bolus significantly improved NP in SCI patients (Table 2) [98]. This shows that baclofen could be a promising NP therapy in the future.
6. Cell Therapy Promoting GABA for SCI-NP
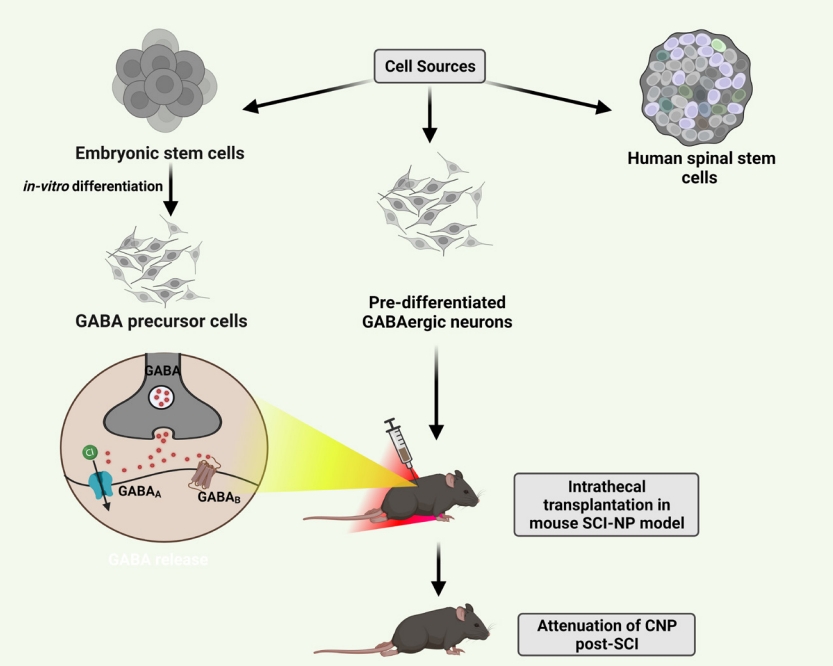
Stem cell therapies avoid deleterious effects of conventional drugs, such as increasing drug tolerance and the development of addiction. Transplanting GABAergic cells in the spinal cord has shown promising effects for NP of nerve injury and SCI [7,99]. When grafted in the paraplegic model of spinal ischemia, human spinal stem cells successfully developed into GABAergic phenotype restoring the inhibitory circuits [100]. In one study, GABAergic precursor cells attenuated NP accompanied hyperalgesia and allodynia in rats when transplanted intrathecal [101]. Correspondingly in other studies, embryonic stem cells from mice, differentiated in vitro into spinal GABAergic neurons, when transplanted postinjury, favored in relieving NP for long term by re-establishing the hypoactive GABAergic tone (Fig. 3, Table 1) [7, 102].
Transplanting selective subpopulations of cells like GABAergic neurons restores the underactive presynaptic inhibition in DH neurons. Transplanting the predifferentiated human neuronal cell line NT2 capable of releasing inhibitory GABA helped reverse NP after quisqualic acid-induced excitotoxic SCI in rats (Table 1) [103]. Similarly, one other study reported reduced overgrooming behavior in SCI rats when transplanted with in vivo predifferentiated GABA immunoreactive cells, for alleviating CNP [104]. The combination therapy of GABAergic neural precursor cells transplantation and intensive locomotor training significantly alleviated the NP symptoms in SCI rats showing mutually beneficial effects [105]. A recent analysis report suggests that GABAergic cells transplantation improves allodynia and hyperalgesia conditions only in rats and not in mice. Additionally, intraspinal transplantation produces more significant effects than intrathecal transplantation. It also suggests that although there are possibilities of genetic defects with genetically modified cells, they are still the best sources for alleviating NP compared to GABAergic cells and stem cells [106].
Studies also report the beneficial effects of bone marrow-derived mesenchymal stem cells (BMSCs) in reducing post-SCI NP and promoting sensory recovery. BMSCs showed effects by inhibiting the activation of MAPK signaling pathway mediated neuronal hyperexcitability [107]. In a case report, BMSCs were administered in a cervical SCI patient, adverse effects including fever, headache, myalgia, and motor dysfunction were observed 48 hours post-transplantation and increased the NP [108]. Although cell therapies with BMSCs are considered safe, it can cause serious adverse effects like hyperthermia and malignant hypertension which requires further investigation [109].
SPASTICITY AND GABA ASSOCIATION
Spasticity and NP are both responsible for affecting the quality of life in patients with SCI by impairing their normal daily activities. They usually have late-onset after the injury and persist for long periods. In patients with the incomplete lesion, spasticity may lead to pain, sleep disorders, or result in loss of motion. Incidence of spasticity after the injury is reported to happen in about 70% of the patients [110]. It is more commonly experienced by quadriplegic patients and in cases with incomplete lesions [111]. Spasticity is defined as ‘a disordered sensorimotor control resulting from an UMN lesion, presenting as intermittent or sustained involuntary activation of muscles [110]. Spasticity is one of the parts of UMN syndrome, which is considered as ‘sensorimotor’ (presents motor responses due to sensory inputs). Although spasticity is considered a detrimental side effect for SCI patients, it is reported to have a beneficial role in locomotory function. So, administration of baclofen even if relieving the spastic symptoms comes with the loss of preserved ambulatory ability in SCI patients [112]. Several mechanisms, including hyperexcitability of motor neurons, the excitability of interneurons, inhibitory GABA-glycinergic decline, and loss of integrity in descending tracts, support the development of spasticity [14,112]. Neuroplasticity brings about alterations in neural circuits, which affect neurotransmission. Although neuroplasticity after SCI promotes locomotor recovery through deafferentation of neurons and neurotrophins, it causes negative effects leading to NP and spasticity after the injury [113]. Activation of plateau potentials due to persistent calcium and sodium inward currents in motor neurons also has a role in spasticity after SCI [114]. By increasing the activity of motor neurons, these persistent inward currents produce uncontrollable spasms that can be provoked even by innocuous stimuli or a muscle stretch. Reduction in expression of cotransporter KCC2 is detected postinjury, triggering EIPSP (reversal potential of glycine receptor and GABAA receptor–mediated inhibitory postsynaptic potentials) depolarization resulting in spasticity below the lesion [115]. Plantier et al. [116] reported the association of Calpain-1 in raising persistent sodium currents and downregulating KCC2 for causing spasticity. Alterations in the transmission of both inhibitory and excitatory tone cause hyperexcitability of neurons, contributing to spasticity. Overall, these mechanisms reduce the motor unit activation threshold and raise the responsiveness to stimuli.
The increase in excitability of motor neurons during spinal ischemia, leading to a spasticity-related increase in muscle tone, is already reported. Baclofen (GABAB agonist) and nipecotic acid (GABA reuptake inhibitor), effectively inhibited the spasticity suggesting the decline in inhibitory GABAergic neurons as a contributing factor to spasticity post-SCI [117]. Progressive loss of cholinergic and GABAergic neurons causing spasticity after sacral SCI is reported in rats. Also, upregulation of downregulated GABAergic receptors was seen after repetitive transcranial magnetic stimulation, which relieved spasticity post-SCI [118]. The global gene expression analysis report of spastic SCI rats signifies the upregulation in the expression of kainate receptors, NMDA receptor complex promoting excitability, and downregulation in gene coding of GABAA receptor subunit isoforms (Gabra1, Gabra5, and Gabrg2) [119]. All in all, inhibitory GABAergic tone insufficiency is a key link involved in spasticity post-SCI.
RESTORING THE INHIBITORY TONE RELIEVES SCI-SPASTICITY
Spasticity post-SCI, although a complication, is beneficial in some aspects requiring no treatment. It may help in the daily activities of SCI patients by increasing stability in sitting and standing, raising muscle bulk, strength, and venous return. Almost 40% of patients find spasticity beneficial. However, depressing the spinal excitability raises motor recovery complications. There is no objective measure for the reference that spasticity should be treated or not, it depends upon achieving the balance of its positive and negative impacts on the patient. Nevertheless, the passive problems of involuntary muscle spasms, persisting pain, and positioning difficulties affecting daily activities need to be treated [112,120]. Several studies target the promotion of inhibitory GABAergic tone in attenuating spastic symptoms (Tables 3, 4).
1. GABAergic Drugs
Several pharmacological agents help in improving spastic conditions (Tables 3, 4). Such interventions classically act to reduce the muscle tone. These drugs, by reducing muscle overactivity, can also attenuate spasticity-mediated NP. Commonly used pharmaceuticals include baclofen, botulinum toxin, tizanidine, whereas diazepam, clonazepam, clonidine, gabapentinoids, skeletal muscle relaxants, cannabinoids, and cyproheptadine are also used, but less commonly [85]. Baclofen, diazepam, tizanidine, and dantrolene sodium are Food and Drug Administration-approved drugs for spasticity of which only baclofen and tizanidine have reported significant efficacy [120]. Of all the options, baclofen is widely used and considered a safer antispastic drug in promoting neurological recovery [121]. Its adverse events are noticed in a dose-dependent way, generally occurring at doses ≥ 60 mg/day when taken orally [120]. When given intrathecal, it has proven to be more potent and efficient than oral baclofen in relieving hypertonia postinjury and avoids its typical side effects of cognitive impairment and sedation [122].
Diazepam by binding GABAA receptors facilitates chloride conductance and promotes presynaptic inhibition, thus attenuating painful spasms in SCI patients. Diazepam and clonazepam carry the risk of dependence and withdrawal syndrome, and on abrupt withdrawal, diazepam can cause seizures [120]. In an open level withdrawal trial, pregabalin worsened the spasticity symptoms in patients on graduate withdrawal protocol [123]. In addition, the development of tolerance and the inability to reach the injury site in effective doses are other causes limiting their use in spasticity. Tolerance to intrathecal baclofen within months after initiating treatment is a complicating factor. Furthermore, some case reports suggest that taking a ‘baclofen holiday’ for 15 days and replacing it with intrathecal morphine infusion is effective in such cases [124]. To counter these complications, other approaches like stem cell therapies are currently being considered.
2. Calcium Channel Blockers
Some studies suggest the use of calcium channel blockers for ameliorating spastic symptoms. Nimodipine, decreased spasticity by blocking persistent inward calcium currents after SCI in spastic mice and rats (Table 3) [114]. A recent study in SCI rats demonstrated that long-term administration of nimodipine reduced pain and spasticity, raised KCC2 expression, spared neurons with the overall promotion of functional recovery. Additional studies in the future with nimodipine might provide further evidence for its use in SCI (Table 3) [125]. Likewise, case reports suggest that ziconotide, by blocking N-type calcium channels, could also relieve spasticity-mediated pain post-SCI. (Table 4) [85,126]. In case reports of baclofen tolerant patients with intractable spasticity, intrathecal ziconotide administration in increasing doses made them spasm free [126]. Sodium and calcium channel blocker tolperisone combined with physical therapy was tested in Chinese SCI patients. It inhibited spasticity with no adverse effects. However, its efficacy was less than baclofen combined with physical therapy (Table 4) [127]. Voltage-gated L-type calcium channel inhibitor 1-(3-chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-(1H,3H,5H)-trione attenuated SCI-induced muscle spasms in mice by inhibiting Cav1.3 subunit specifically. This suggests Cav1.3 channel-mediated excitability inhibition could be a promising target for relieving SCI-spasticity in the near future [128].
3. Cell Therapy
Researchers are now focusing more on cellular approaches for treating SCI-spasticity. Cells like bone marrow-derived stromal cells can positively acquire GABAergic type and migrate towards the injury site preserving the neuronal survival and regeneration in the spinal cord [129]. The study conducted by Gong et al. [130] transplanting the GABAergic neurons into the rats after their successful differentiation from progenitors established the role of inhibitory GABAergic neurons in relieving spasticity (Table 3). Intraspinal grafting of human spinal cord-derived stem cells (NSI-566RSCs line) had promising effects on the functional recovery of SCI rats. It attenuated spasticity and NP symptoms by developing putative synapses between GABAergic neurons and the host’s interneurons and motor neurons (Table 3) [131]. Human trials have demonstrated the safety of neural stem cell transplantation post-SCI. However, the major hurdles include lack of proper differentiation, glial scarring, hostimmune reactions, and risk of tumorigenesis [132].
4. KCC2 Cotransporter Enhancers
The cotransporter KCC2 required to maintain inhibitory GABAergic tone is downregulated post-SCI contributing to spasticity development [115]. Consequently, current studies also focus on upregulating KCC2 levels to increase chloride extrusion to reduce spasticity. The antipsychotic drug prochlorperazine, showed increased KCC2 expression to relieve spasticity and the antispastic effect was comparable to baclofen (Table 3) [133].
NEUROPATHIC PAIN AND SPASTICITY INTERLINKED
Maladaptive neuroplasticity post-SCI is the reason for a number of negative consequences, such as spasticity and NP [113]. Both NP and spasticity are linked with common causal mechanisms like hypoactive inhibitory tone and neuronal hyperexcitability, which reduce the threshold and enhance responses of spinal neurons to non-painful stimuli. A cross-sectional survey reported that the prevalence of spasticity is more common with NP patients when compared to patients with no pain [134]. The key mechanism connecting both is KCC2 downregulation. The only difference is the reduction in expression of KCC2 in NP is observed mainly in DH neurons, whereas in the case of spasticity, its expression is reduced primarily in motor neurons [22].
Many approaches target SCI-induced NP and spasticity simultaneously. BTX-A shares antispastic and antinociceptive properties [75,85]. The first-line agent used for NP, gabapentin, also suppresses spasticity (Table 3) [135]. Similarly, cannabinoids that modulate the neuronal hyperexcitability and neurotransmitter release, can attenuate NP and spasticity. However, their uncertain efficacy in relieving spasticity requires more research and could be confirmed from the results of ongoing trials [136].
CONCLUSION
The secondary phase of SCI brings more distressing complications as a result of alterations at cellular, molecular and genetic levels. Changes occurring due to damage alter normal synaptic transmissions and causes neuroplasticity contributing to the development of CNP and spasticity post-SCI. Their prevalence post-SCI is high and remains refractory to the conventional approaches. Current pharmacological therapies lack effectiveness and come with a wide range of side effects. Also, targeting complex pain mechanisms itself becomes challenging. Thus, defining these mechanisms becomes a need to develop new approaches. Newly synthesized drugs targeting the GABAergic system to relieve CNP bring the possibility of investigating them in SCI. Similarly, repurposing of GABAergic drugs for the chronic period in SCI, gene therapies designed to promote GABAergic tone, and stem cell transplantation, primarily GABAergic neurons, are some of the recent advancements in CNP and spasticity post-SCI that provide insight to the ongoing research.










")













