INTRODUCTION
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare and highly aggressive embryonal tumors of the central nervous system (CNS) that were first defined by Rorke et al. in 1996 [1]. Male predominance is observed, and the male to female ratio ranges from 1.3 to 1.5 [2]. Although various ages at diagnosis have been reported from studies, the median age at diagnosis ranges between 6 and 12 months [3,4]. Most cases occur in patients less than 3 years old [3]. While AT/RTs represent 1.6% of all CNS tumors diagnosed in patients less than 19 years old, the percentage has been reported to be 10.1% in patients under 1 year old [3]. AT/RTs predominantly occur in infratentorial and supratentorial locations but rarely in the spinal canal; AT/RTs in the spinal canal account for 2% to 4% of all AT/RTs [3,5]. Biallelic SMARCB1 (INI1, SNF5, BAF47) or rare SMARCA4 (BRG1) inactivation, encoding core subunits of the SWI/SNF chromatin-remodelling complexes, has been shown to be a common genetic alteration and the central event in AT/RTs [2,6]. The five-year overall survival (OS) rates of patients with AT/RTs are between 15% and 50%, even though these rates have recently improved due to multimodal treatment approaches [5]. Older age, radiotherapy, gross total resection (GTR) and absence of metastasis are most consistently related to a better prognosis [7]. Intensive chemotherapy has been validated as an efficient treatment modality for AT/RTs [8].
Due to the uncommon incidence of AT/RTs in the spinal canal, there is no consensus concerning a standard treatment regimen for these tumors, and prognostic factors remain unclear. The aim of this systematic review was to summarize and analyze the demographics and clinical presentations, pathological characteristics, treatment interventions and prognosis of AT/RTs in the spinal canal and to validate potential prognostic factors in patients with this category of tumor.
MATERIALS AND METHODS
1. Literature Search
We retrieved spinal AT/RT-related articles from the PubMed, MEDLINE, and Embase databases. A literature search for publications between 1980 and July 2023 with the following key words was conducted: “atypical teratoid/rhabdoid tumor,” “rhabdoid tumor,” “spine,” “spinal,” “spinal neoplasm”, and “spinal cord neoplasm.”
2. Study Inclusion and Analysis
We included articles adhering to the following criteria: published in English, some or all tumors located in the spinal canal and AT/RTs with SMARCB1-deficient expression validated by pathological examination. Articles were excluded if AT/RT in the spinal canal was not the primary tumor. In all, 598 potentially relevant articles were retrieved. The quality of the included studies was assessed by the Joanna Briggs Institute Checklist for Case Reports and Case series to control the risk of bias. This checklist includes 8 items for case reports and 10 items for case series. The answers for the checklist can be rated as 1 of the 4 options: yes, no, unclear, or not applicable. For case reports, only if the overall scores reached at least 6 points could the studies be included. For case series, overall scores of at least 7 points were required. Two authors assessed the title and abstract of each article and then reviewed full texts meeting the inclusion criteria. A consensus regarding article eligibility was reached according to the inclusion and exclusion criteria. Data regarding clinical presentation, radiological features, pathological characteristics, comprehensive treatment regimens, and prognosis were further analyzed. Progression-free survival (PFS) was defined as the duration from diagnosis to first progression (recurrence or metastasis), death resulting from any cause or last follow-up. OS was defined as the duration from diagnosis until death or last contact. PFS and OS curves were estimated by Kaplan-Meier analysis. The log-rank test and Breslow test were utilized to evaluate the intergroup differences in the single-factor analysis. All parameters with significant differences in the univariate Cox regression analysis were further analyzed in the subsequent multivariate analysis. The hazard ratios and 95% confidence intervals were calculated to validate the independent prognostic factors related to PFS and OS in patients with primary spinal AT/RTs. A p-value of < 0.05 was defined as a significant difference. Statistical analyses were performed with IBM SPSS Statistics ver. 25.0 (IBM Co., Armonk, NY, USA).
RESULTS
1. Patient Demographics and Clinical Characteristics
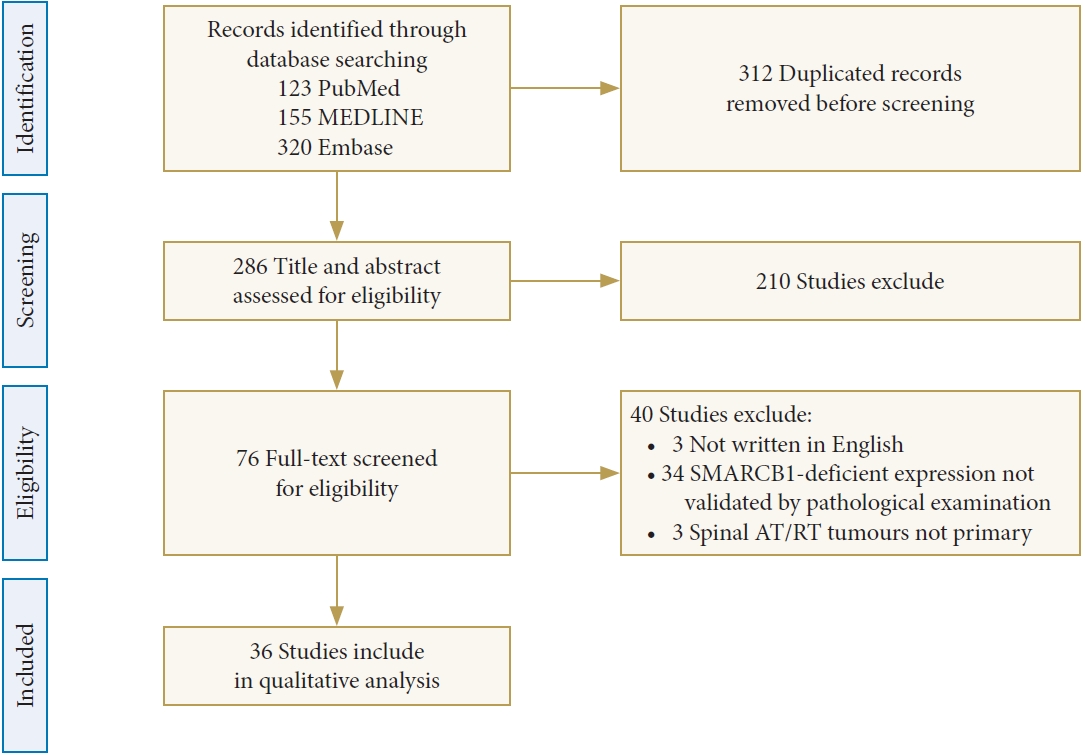
To identify potential prognostic factors for spinal AT/RTs, we explored the PubMed, MEDLINE, and Embase databases using relevant keywords. After the exclusion of 562 articles, 36 articles met the eligibility criteria and described in detail 58 patients with primary spinal AT/RTs from 1980 to July 2023 that were validated by pathological examination (Fig. 1) [7,9-43]. Spinal AT/RT tumors in 7 cases (12.3%) invaded and broke through the dura or leptomeninges. Most patients (30 cases, 52.6%) had intradural and extramedullary tumors, and 20 patients (35.1%) and 14 patients (24.6%) had intramedullary and extradural tumors, respectively. Eleven cases (19.0%) exhibited extension into the intervertebral foramen, and 8 cases (13.8%) demonstrated infiltration of nerve roots on neuroimaging. The male to female ratio was close to 1 (29:28), and the median age at diagnosis was 4 years (range, 0.3–65.0 years). The majority of the tumors were located in the lumbar and thoracic segments (35 cases, 60.3%; 32 cases, 55.2%, respectively), and the rest were located in the cervical and sacral segments (19 cases, 32.8%; 12 cases, 20.7%, respectively). Among the 58 cases, 31 cases (53.4%) showed involvement of more than one spinal segment. The most frequently reported clinical presentations were pain and extremity weakness, with a median clinical history duration of 1 month (range, 0.2–29.5 months) in 33 patients based on the longest symptomatic duration of each of these patients. The median maximum diameter of the tumors in 19 patients was 5.8 cm (range, 1.4–14.3 cm), with 8 tumors (61.5%) and 5 tumors (38.5%) showing obscure and clear boundaries, respectively. The presentation of T1-weighted imaging (T1WI) and T2-weighted imaging (T2WI) signals was highly variable; nevertheless, 36 tumors (62.1%) had enhancement, and heterogeneous enhancement was more common in 14 of these tumors (38.9%). There were 14 cases (25.0%) of leptomeningeal dissemination at preoperative diagnosis and 12 cases (21.4%) of leptomeningeal dissemination at postoperative diagnosis. Moreover, 1 case (1.8%) of leptomeningeal dissemination was confirmed by autopsy. Apart from the overlapping cases, 23 cases (41.1%) of leptomeningeal dissemination were found. Moreover, 8 patients developed hydrocephalus during the clinical course. The demographic and clinical features of these patients are summarized in Table 1.
2. Treatment Modalities
Forty-eight patients underwent surgical treatment: subtotal resection (STR) in 31 patients (64.6%) and GTR in 17 patients (35.4%). Nine patients were treated with secondary operations after disease progression. Additionally, 30 patients were treated with postoperative radiotherapy, and 39 patients were treated with postoperative chemotherapy. Fourteen of 39 patients (35.9%) treated with chemotherapy underwent intrathecal chemotherapy.
3. Pathological Characteristics
Immunohistochemical examinations indicated that vimentin was positive in 24 of all patients examined for vimentin, S100 in 3 of 10, desmin in 1 of 16, glial fibrillary acidic protein in 3 of 19, epithelial membrane antigen in 33 of 36, cytokeratin in 24 of 33, anti-smooth muscle antibody in 11 of 17, neurofilament protein in 6 of 7, CD99 in 6 of 12 and synaptophysin in 4 of 14. The loss of SMARCB1 protein was validated by immunohistochemical examinations in all cases, and the index of Ki-67 labelling ranged from 10% to 90% in 14 cases. Furthermore, tumors in 14 cases (24.1%) demonstrated haemorrhage and necrosis. The treatment modalities and pathological characteristics of these patients are summarized in Table 2.
4. Follow-up and Prognosis
The mean follow-up duration was 18.1 months, with a range of 0.6–93.0 months. Nine patients suffered from recurrence (9 of 33, 27.3%), and 19 patients developed metastasis (19 of 37, 51.4%), including 6 cases (6 of 19, 31.6%) with intracranial metastasis and 1 case with extra-axial metastasis. Twenty-eight patients passed away during the follow-up period (28 of 51, 54.9%). The 2-year PFS and OS rates were 39.5% and 42.8%, respectively. The follow-up and prognosis of these patients are summarized in Table 3.
5. Statistical Data Analysis
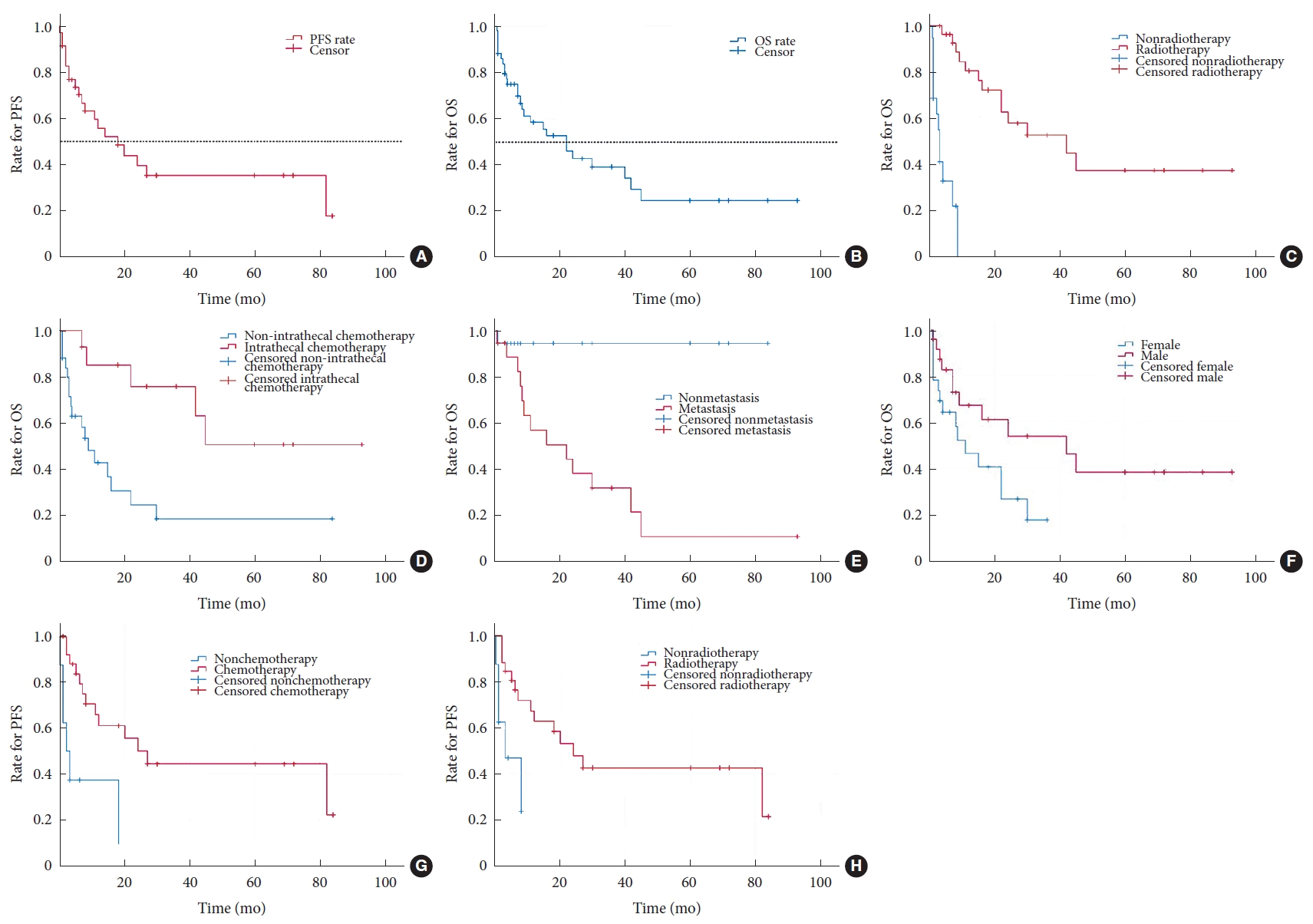
A summary of the descriptive statistical data is shown in Table 4. The median PFS and OS were 18 and 22 months, respectively, as demonstrated by the Kaplan-Meier curves in Fig. 2A and B, respectively. Patients who received radiotherapy (log-rank test: p < 0.001; Breslow test: p < 0.001) or intrathecal chemotherapy (log-rank test: p = 0.003; Breslow test: p = 0.003) had a longer OS than those who did not receive radiotherapy or intrathecal chemotherapy according to Kaplan-Meier analysis (Fig. 2C and D, respectively). In addition, a significant difference in OS for the metastasis (log-rank test: p = 0.002; Breslow test: p = 0.020) and sex (log-rank test: p = 0.040; Breslow test: p = 0.056) groups was observed based on Kaplan-Meier analysis (Fig. 2E and F, respectively); this finding indicated that patients without metastasis and males had a better OS than those with metastasis and females, respectively. A similar analysis indicated that patients receiving chemotherapy (log-rank test: p = 0.001; Breslow test: p = 0.001) or radiotherapy (log-rank test: p = 0.007; Breslow test: p = 0.006) exhibited a better PFS (Fig. 2G and H, respectively). Multivariate Cox regression analysis revealed that patients treated with chemotherapy (p = 0.010) or radiotherapy (p = 0.034) had a lower risk for disease progression than those without chemotherapy or radiotherapy. A homologous analysis found a lower mortality risk for patients treated with intrathecal chemotherapy (p = 0.042). The statistical analysis results of these patients are summarized in Table 5.
DISCUSSION
1. Patient Demographics
AT/RTs are highly invasive and aggressive CNS embryonal tumors that are most frequently located in supratentorial and infratentorial locations and rarely in the spinal canal [3]. AT/RTs occur in 1.6% of patients under 19 years old diagnosed with CNS tumors, and spinal AT/RT cases account for 2%–4% of validated AT/RT cases [3,5]. AT/RTs mainly affect children younger than 3 years with a slight male predominance. AT/RTs in adults are rare; nonetheless, accumulated cases have demonstrated that AT/RTs in the sellar region exclusively arise in adult females, implicating biological differences from other AT/RTs [44]. A study comprising 6 cases of sellar AT/RTs by Nakata et al. [44] demonstrated an age range from 21 to 69 years and a significant female predominance in which all patients were female. However, there was no significant sex predominance detected in our research. Nonetheless, a significant difference was observed in the sex group, which indicated that males had a better prognosis than females. The median age of the 44 patients in our study was 4 years, with a range of 0.3–65.0 years, which was generally consistent with prior research. Younger patients at the time of diagnosis of AT/RTs have been identified as having a worse prognosis than older patients [5,45,46]. Although there is a tendency for improved survival in patients 4 to 17 years old compared with patients aged under 1 to 2 years at diagnosis, our research did not reveal a significant difference in survival based on age [47].
2. Neoadjuvant Therapy
While GTR could improve the prognosis of patients with AT/RTs, it is often difficult to perform GTR at primary surgery due to the large size and abundant vascularity of spinal AT/RTs [46]. Previous studies indicated that GTR is achieved in 30%–50% of AT/RTs [8,46,47]. The GTR rate in our study was merely up to 35.4%. To boost the GTR rate of spinal AT/RTs, neoadjuvant chemotherapy is necessary, which may diminish not only the size but also the vascularity of tumors and facilitate subsequent resection [48]. Unfortunately, no case in our research was observed to utilize neoadjuvant therapy. Even though the utilization of neoadjuvant therapy in AT/RTs is infrequent, a study of a small cohort conducted by Ishisaka et al. [48] in which ICE or modified IRS-III regimens were administered to patients preoperatively was noted and indicated that neoadjuvant therapy could diminish the volume and vascularity of tumors, but no improvement in prognosis was observed.
3. Surgery and Surgery-Related Complications
Due to a lack of large amounts of clinical data on primary spinal AT/RTs, there are currently no effective and mature treatment regimens. An aggressive surgical approach is the mainstay of treatment for spinal AT/RTs. A study by Hilden et al. [46] indicated that GTR could improve the PFS of patients and the prognosis of AT/RTs. GTR was achieved in 35.4% of patients in our research. Maximal-degree resection of the primary tumor has been associated with improved PFS and OS [47]. Significant differences in OS and PFS for the administration of surgery or the extent of tumor resection were not observed based on Kaplan-Meier analysis. Nevertheless, patients who underwent surgery had longer median OS (24 months vs. 7 months) and PFS (20 months vs. 14 months) than did those who did not undergo surgery, and patients who underwent GTR had longer median OS (42 months vs. 15 months) and PFS (20 months vs. 11 months) than did those who underwent STR. Therefore, an aggressive surgical approach is recommended to achieve GTR, which could alleviate symptoms, sufficiently reduce tumor volume, obtain tumor specimens for pathological examinations and form the basis for subsequent treatment. En bloc tumor resection with negative margins is often regarded as an effective method of achieving long-term PFS or OS in some cases of malignant spinal neoplasms [49]. Only one case in our study was treated with en bloc resection and there were no reports to be found regarding the extent of surgical margins. Because of the malignant biological behavior of spinal AT/RTs, the tumors commonly invade nerve roots, vertebral arteries and cervical spinal cord. Obtaining a negative surgical margin is difficult because damage to the aforementioned structures during aggressive tumor resection will cause severe complications and even death. Nonetheless, pursuing a goal of negative surgical margins would benefit the patients greatly under the premises of extensive presurgical planning, multidisciplinary surgical teams with rich experience managing these tumors and early-stage input of rehabilitation physicians following aggressive resections [50]. Surgical-related complications depend on both aggressive surgical resection and the patient’s medical background. We did not observe any early (less than 30 days postoperatively) complications in our study but observed one patient with chronic (more than 30 days postoperatively) complications and progressive thoracolumbar kyphosis 5 years after emergent T9-L3 laminectomies for STR. Twenty-three patients in our study did not develop neurological deficits or deterioration of vital status in the early postoperative stage. Given the malignant biological behavior of these tumors, after extensive discussion with the patients and their respective families, surgery should not be compromised to avoid neurological deficits with the aim of saving life when the tumors, such as those located in the cervical segments, endanger the lives of patients. Conversely, if the tumors cannot endanger the patient’s life shortly, surgery should be performed with a view to avoiding the development of neurological deficits and providing the patient with optimum long-term tumor control. In 23 available cases with disease progression, we observed 9 patients undergoing secondary surgery. However, there were no significant differences demonstrating that secondary surgery could improve the prognosis of patients with disease progression. Considering spinal AT/RTs as systemic diseases and highly malignant tumors, multiple operations could increase the risks of drop metastasis and severe neurological deficits and even cause the death of patients. Meanwhile, chemotherapy and radiotherapy have shown progress in helping to improve the prognosis of spinal AT/RTs. Thus, it might be more prudent to choose secondary surgery treatment when the patient suffers a relapse.
4. Ki-67 and Molecular Cytogenetics
Of the immunohistochemical markers, Ki-67 antibody has been validated as providing the most valuable approach for evaluating the cell proliferation extent of CNS neoplasms [51]. Ki-67 was recorded in 14 cases, ranging from 10% to 90%. No significant difference was observed in our study probability due to the limited Ki-67 data. Thus, efforts to obtain more clinical data are warranted in the future. SMARCB1 has been validated as a genuine tumor suppressor gene in genetically engineered mouse models; biallelic inactivation of SMARCB1 located in chromosome band 22q11 [2]. is the most ubiquitous genetic alteration in AT/RTs [2,52]. SMARCB1 inactivation has a variety of causes such as deletions, loss of heterozygosity and mutations, which result in the mutation of subunits of SWI/SNF complexes encoded by the SMARCB1 gene, the central propulsion event in the development of AT/RTs [2].
5. Role of Chemotherapy
Our analysis demonstrated that the utilization of chemotherapy and intrathecal chemotherapy could be potential positive prognostic factors in spinal AT/RTs, as validated by Kaplan-Meier analysis and multivariate Cox regression analysis. The patients receiving intrathecal chemotherapy had improved OS and a lower mortality risk, which might be associated with a high incidence of leptomeningeal dissemination of spinal AT/RTs. High-dose chemotherapy (HDC) combined with stem cell rescue (SCR) is considered an effective regimen for AT/RT and allows radiotherapy to be deferred in very young children to prevent neurocognitive deficits [53]. The Head Start and CCG99703 regimens represented the first generation of such studies, from which there were reports of favorable prognosis for a proportion of patients with HDC/SCR. Slavc et al. [54] reported a series with an improved 5-year OS of 100% in 9 patients undergoing an intensive multimodal regimen consisting of a dose-dense regimen combined with intrathecal chemotherapy followed by HDC/SCR. Importantly, as the largest prospective research for AT/RTs, the ACNS0333 trial produced results that confirmed HDC/SCR as an important regimen in AT/RTs.
6. Role of Radiotherapy
In terms of therapeutic regimens for more frequent embryonal CNS tumors, radiotherapy is deemed to represent an important regimen for spinal AT/RTs. While the utilization of radiotherapy in spinal AT/RT patients remains controversial due to severe neurocognitive side effects in young children, Buscariollo et al. [47] found that younger age had an even stronger relationship with a more favorable prognosis when radiotherapy was utilized as the initial treatment. In one case reported by Yano et al. [33] in our research, it was noted that a patient, treated with GTR and intensive systematic and intrathecal chemotherapy with autologous bone marrow transplantation upon diagnosis at 1.75 years of age, received radiotherapy to the local tumor bed and craniospinal axis after 1 year and maintained complete remission without neurological deficits at the last follow-up at 4 years of age. Decision-making regarding the benefit-risk of radiotherapy use for treating children with AT/RTs remains difficult. Nevertheless, such cases highlight that dynamic and individual radiotherapy designs might be necessary for some children in the early-stage of spinal AT/RTs. Conversely, for very young patients with relapse, the survival profit may outweigh the possible hazard, and focal irradiation of small volumes can be performed to minimize side effects [55]. Stereotactic radiosurgery (SRS) is a potential therapeutic option compared with conventional radiation therapies in that SRS enables a highly conformal and large dose radiation to be delivered to a localized tumor tissue while minimizing radiation exposure in normal tissue. The safety of SRS for treating patients with spinal tumors has been confirmed by multiple reports [56-59]. A study by Benzil et al. [57] revealed that SRS was safe with minimal risk of complications and provided effective pain relief in the treatment of spinal tumors, but did not indicate obvious improvement of prognosis. Another study by Shin et al. [56] demonstrated effectiveness in tumor control and the improvement of neurological function for spinal metastases. Nonetheless, the long-term survival of patients undergoing SRS is still dismal. Spina et al. [58] conducted a systematic review regarding SRS for the management of AT/RTs and found that local tumor control was achieved in 66.7% (8 of 12) of cases, among which 4 cases were alive with a mean survival time of 61.3 months. SRS was administered in one patient with intracranial metastasis postoperatively, and the patient died 2.5 years after diagnosis in our study. Despite the lack of long-term survival improvement evidence, potential benefits of utilizing SRS in the management of spinal AT/RTs as an initial irradiation regimen are avoiding the increased risks of radiotherapy-related neurocognitive and endocrine toxicity, alleviating symptoms and improving local tumor control for young children with spinal AT/RTs. These relationships among age, irradiation utilization, and magnitude of the relevant difference in survival for spinal AT/RTs warrant validation in clinical trials. Compared with historical therapies, the ACNS0333 regimen conducted by Reddy et al. [8], utilizing high-dose methotrexate induction with HDC/SCR consolidation combined with involved-field and age-adapted timing radiotherapy, dramatically improved the survival of patients with AT/RTs and might provide a promising reference for spinal AT/RT treatment.
7. Clinical Outcome and Patterns of Failure
Of the 50 patients included in our analysis, the median OS was 22 months, as calculated by Kaplan-Meier analysis, which was longer than the range of 6 to 18 months in previously published studies on AT/RTs [47,60]. Spinal AT/RTs in our research revealed that the characteristics of surrounding invasion and metastasis on neuroimaging were ubiquitous, including multiple spinal-segment involvement, invasion and breaking through leptomeninges or dura, extension into interverbal foramens, infiltration of nerve roots and presentation of obscure boundaries at diagnosis. The frequency of tumor metastasis was 51.4% in 37 patients with available information, and this value was higher than the range (i.e., 20% to 40%) reported in prior studies regarding AT/RTs [4]. Furthermore, 6 patients developed intracranial metastasis, and one patient developed extra-axial metastasis. Our research indicated that metastasis was associated with poorer OS. A higher incidence of metastasis and poorer survival of patients with spinal AT/RTs indicated that tumors in this category were more aggressive and invasive than cranial AT/RTs. Approximately 25%–35% of AT/RT patients with germline mutations in SMARCB1 are diagnosed in generally younger patients than those without SMARCB1 germline mutations, who show a predisposition to concurrent tumors and more extensive disease [2,8]. Thus, patients, particularly at an earlier age, should undergo whole-body examinations to detect potential extra-axial lesions, if pathological examination validates the diagnosis of spinal AT/RTs. Postoperative neuroimaging examination including the whole neural axis is warranted to find early-stage cranial and spinal metastasis and perform timely intervention in spinal AT/RT patients. Moreover, we assessed 14 cases showing signs of leptomeningeal dissemination on neuroimaging at preoperative diagnosis and 12 cases showing leptomeningeal dissemination after operation administration. Additionally, one case of leptomeningeal dissemination was confirmed by autopsy. Apart from the overlapping cases, 23 cases of leptomeningeal dissemination were found. Leptomeningeal dissemination accounts for one-third of AT/RTs, and its prognosis is poor, with a median survival of 1–4 months [61,62]. A study by Calandrelli et al. [62] described 4 patients with leptomeningeal dissemination, accounting for 40% of all pediatric patients at diagnosis. Another study on AT/RTs by Dardis et al. [63] demonstrated that 12 cases of leptomeningeal dissemination accounted for 24% of 50 cases and that adults with leptomeningeal dissemination had a grave prognosis. While survival analysis indicated that leptomeningeal dissemination was not a prognostic factor for OS in our research, our review of clinical findings indicated that spinal AT/RTs adjacent to cerebrospinal fluid (CSF) resulted in a high incidence of leptomeningeal dissemination and the presence of metastasis. Therefore, the early-stage detection of leptomeningeal dissemination in spinal AT/RTs should be highlighted for its high incidence to guide treatment regimens and evaluate prognosis. Moreover, 8 cases were identified as hydrocephalus during the clinical course. Despite no significant differences between the hydrocephalus and nonhydrocephalus groups, the median OS of patients with hydrocephalus was shorter than that of patients with nonhydrocephalus (16 months in 7 patients vs. 30 months in 43 patients). Neoplastic leptomeningeal dissemination, extension of tumors in high-level spinal segments and intracranial metastasis to block CSF pathways might be mechanisms for the development of hydrocephalus in spinal AT/RTs [64].
8. Limitations
This review is limited by undetailed and incomplete data as well as small sample size. Because of the importance of cell proliferation, more information regarding Ki-67 should be collected and analyzed to assist clinicians in evaluating the prognosis of spinal AT/RTs. More regimens such as neoadjuvant therapy, SRS and secondary surgery should be applied in a large-scale study to further validate their efficacy. Owing to the lack of DNA methylation profiling and gene expression profiling information, we could not classify spinal AT/RTs into 3 molecular subtypes according to the fifth edition of the World Health Organization classification of CNS tumors. Molecular analysis, such as whole-genome or whole-exome sequencing, should be conducted in more cases of spinal AT/RTs. Structural variants, mutations and DNA methylation profiling should be screened, collected, and analyzed in the future to explore pathogenesis and potential targeted agents against spinal AT/RTs.
CONCLUSION
Primary spinal AT/RTs are aggressive malignant tumors with a dismal patient survival rate, even after intensive treatment regimens. Metastasis and female sex were associated with poorer prognosis. As the basis of spinal AT/RT therapeutic regimens, surgical resection, particularly maximal-degree resection, could prolong the median OS and PFS of patients. Chemotherapy might defer the time of disease progression and reduce the risk of disease progression. We also found that intrathecal chemotherapy could extend the OS of patients. Radiotherapy was validated as a feasible and effective therapeutic regimen that could prolong the OS and PFS of patients and lower the risk of disease progression. However, further larger-scale prospective studies of spinal AT/RTs are warranted to confirm these findings and explore additional prognostic factors and specific treatment regimens.